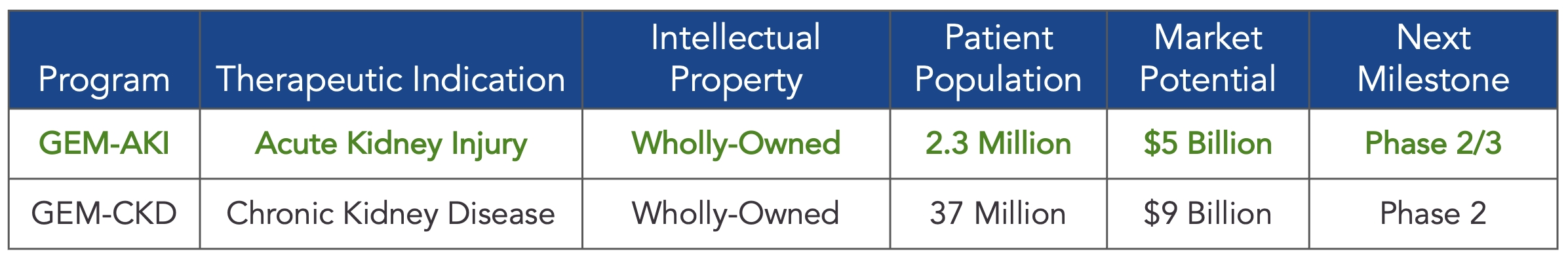
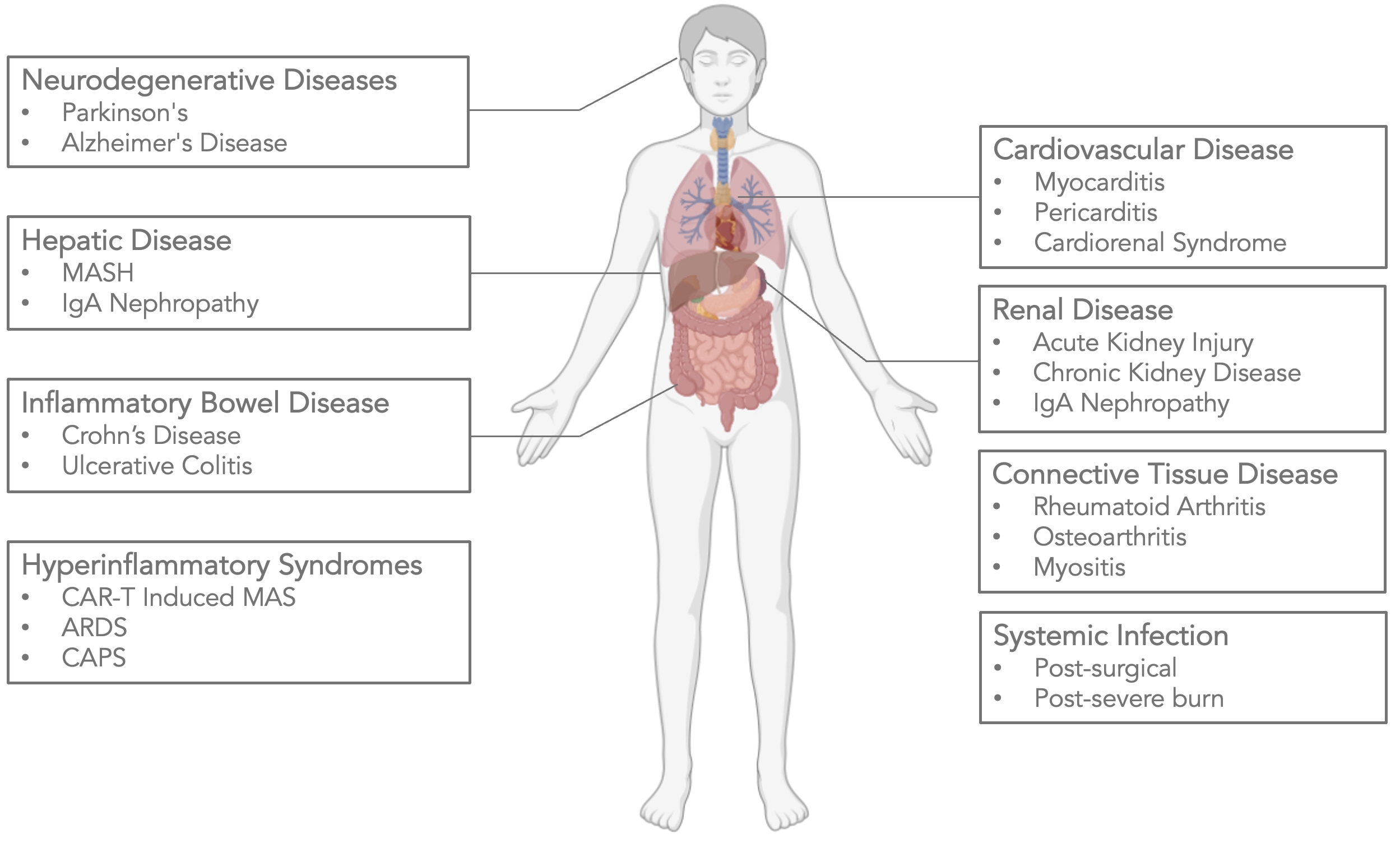
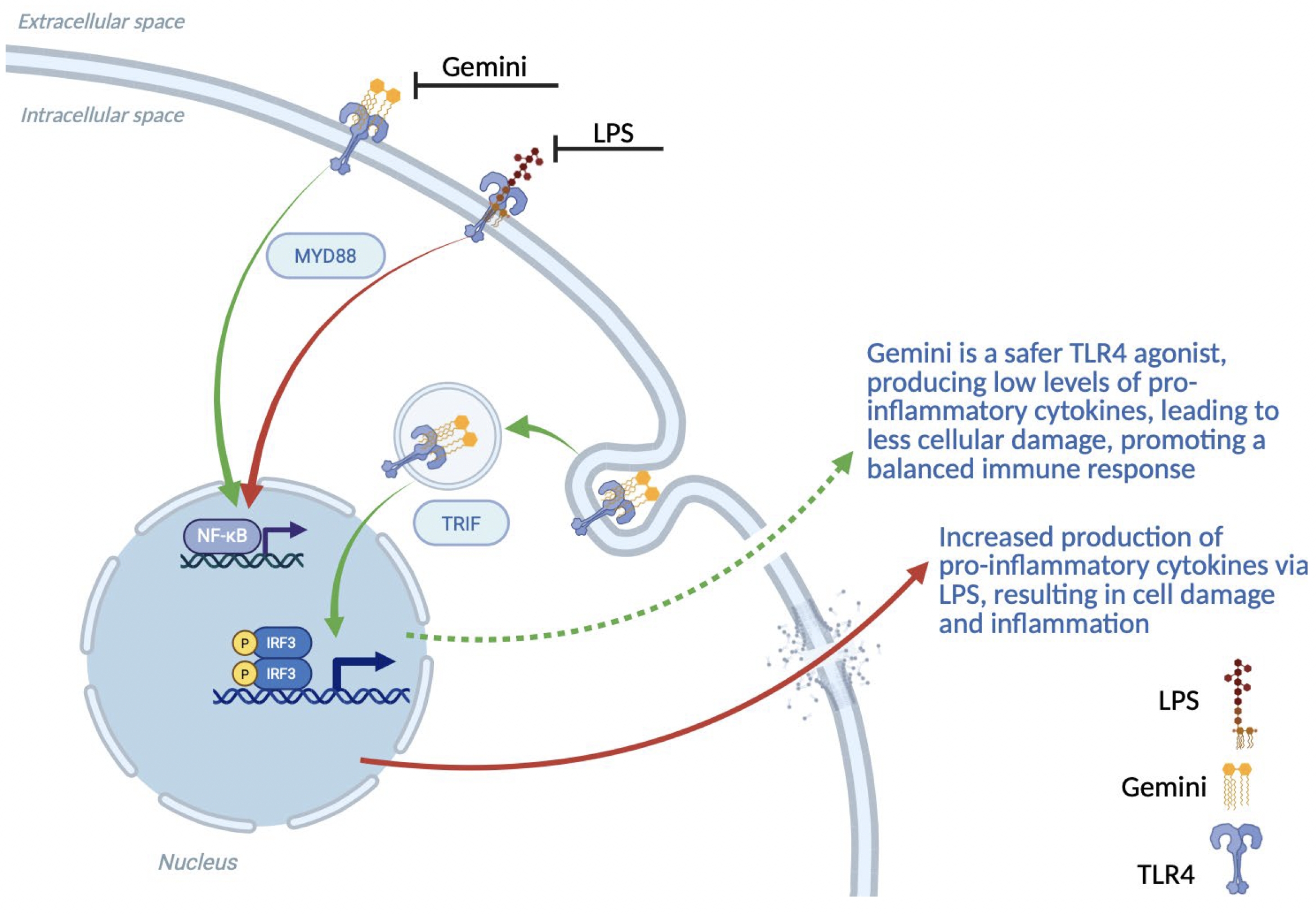
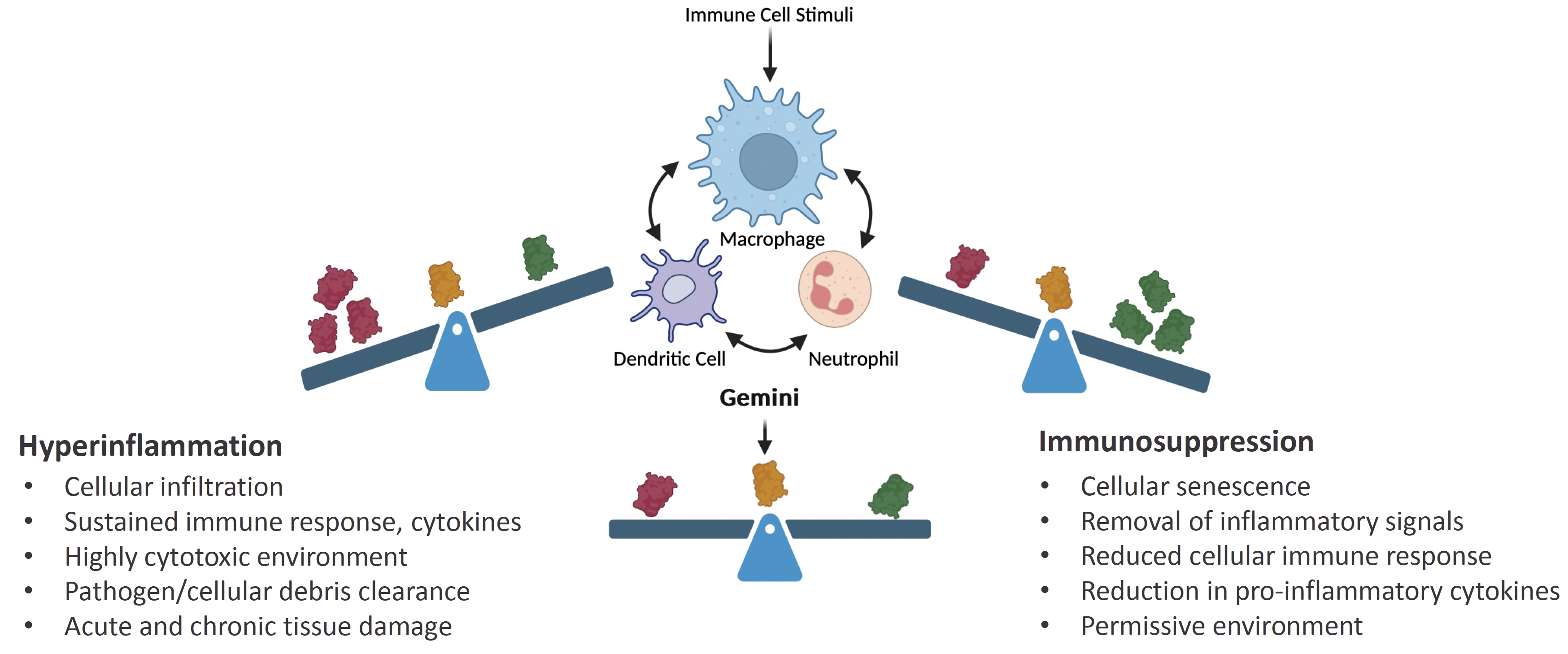
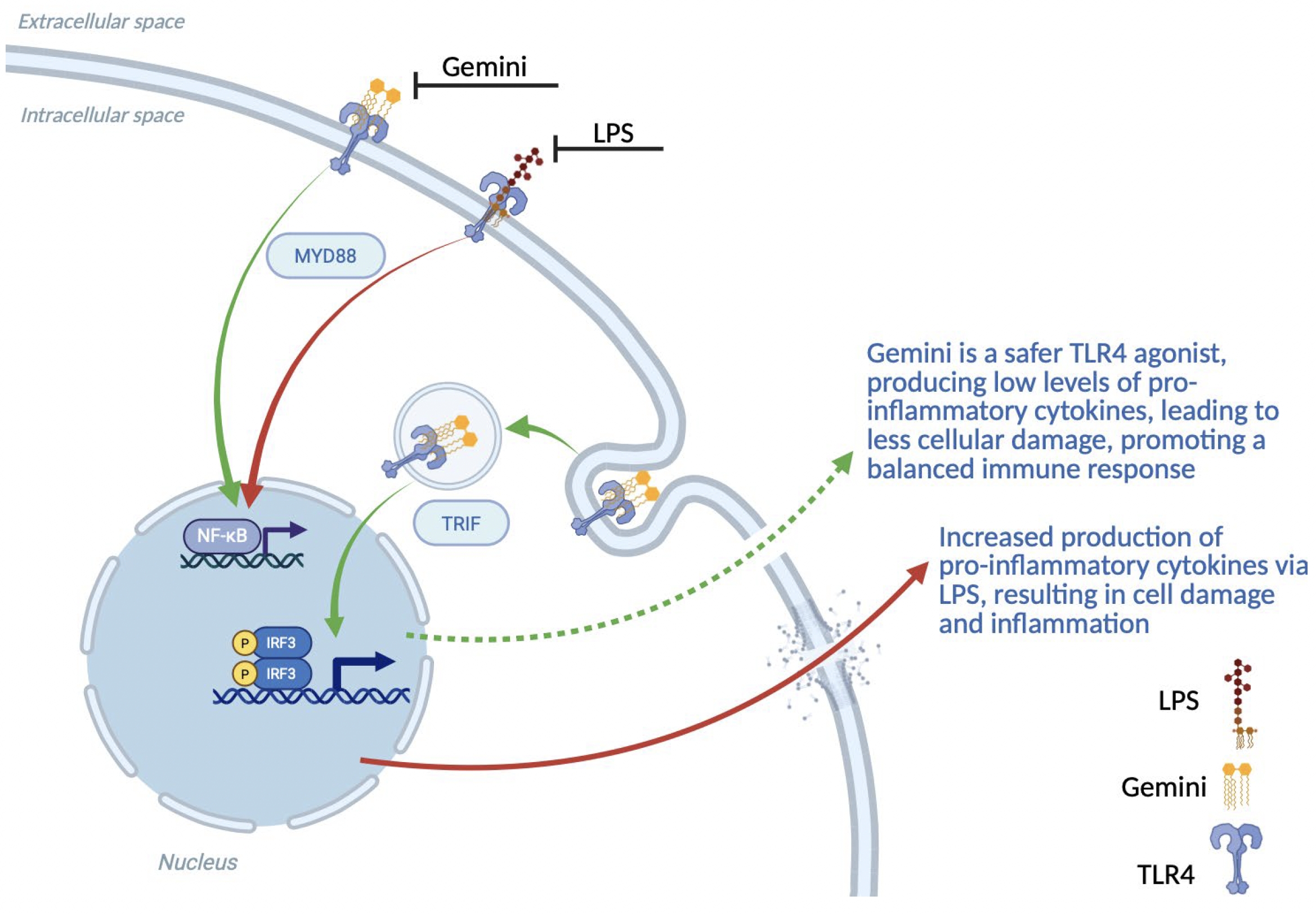
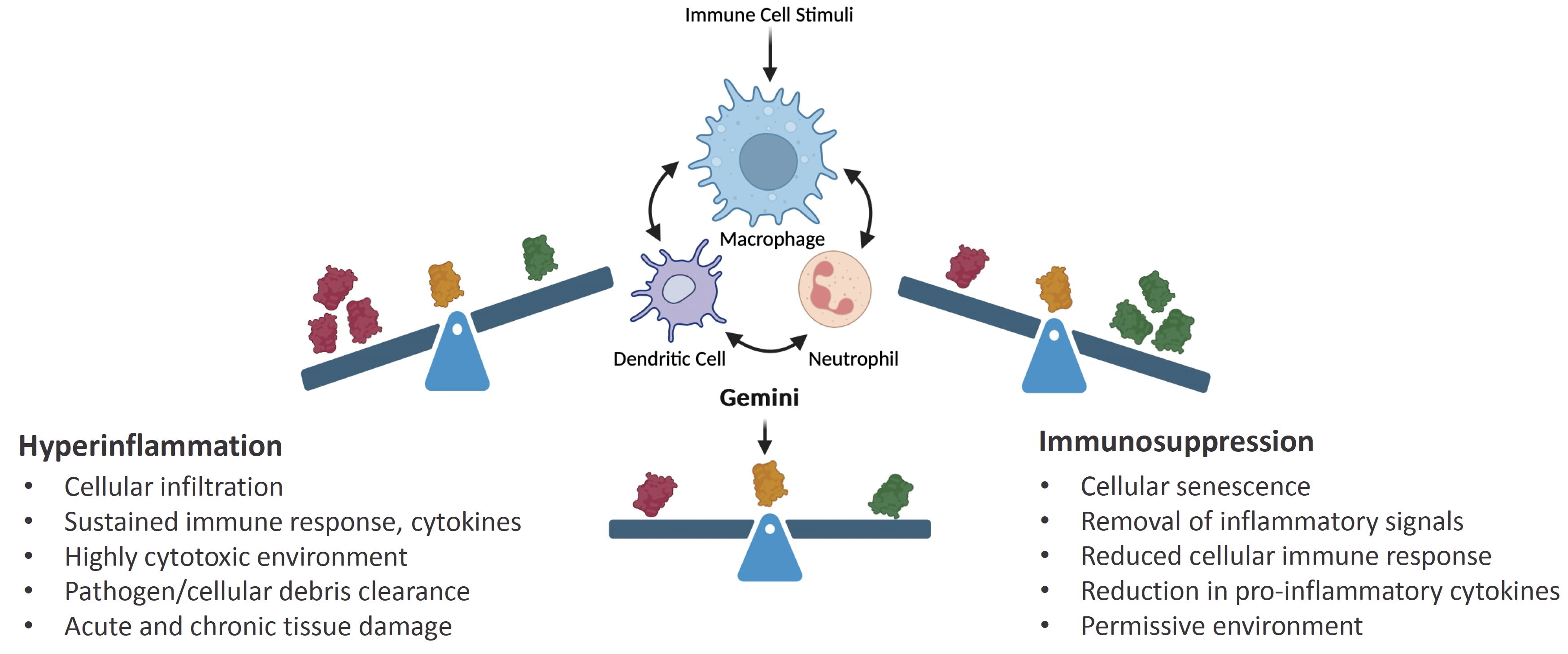
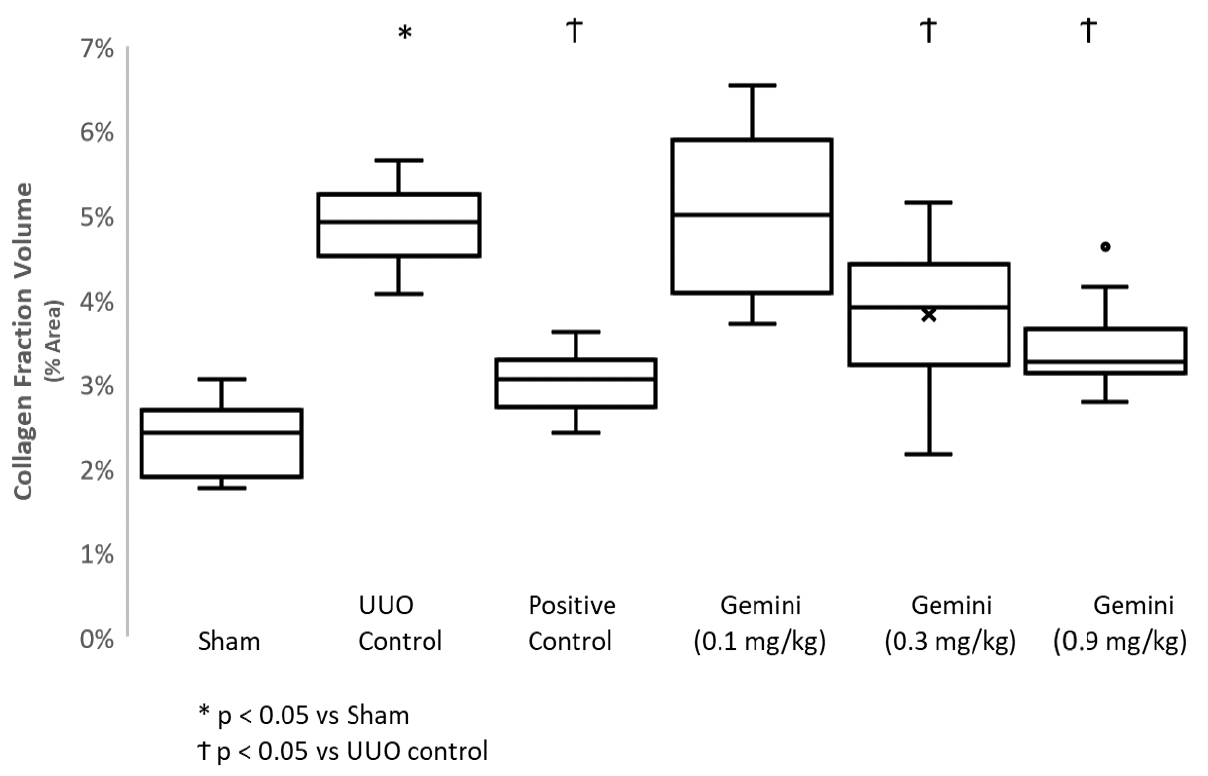
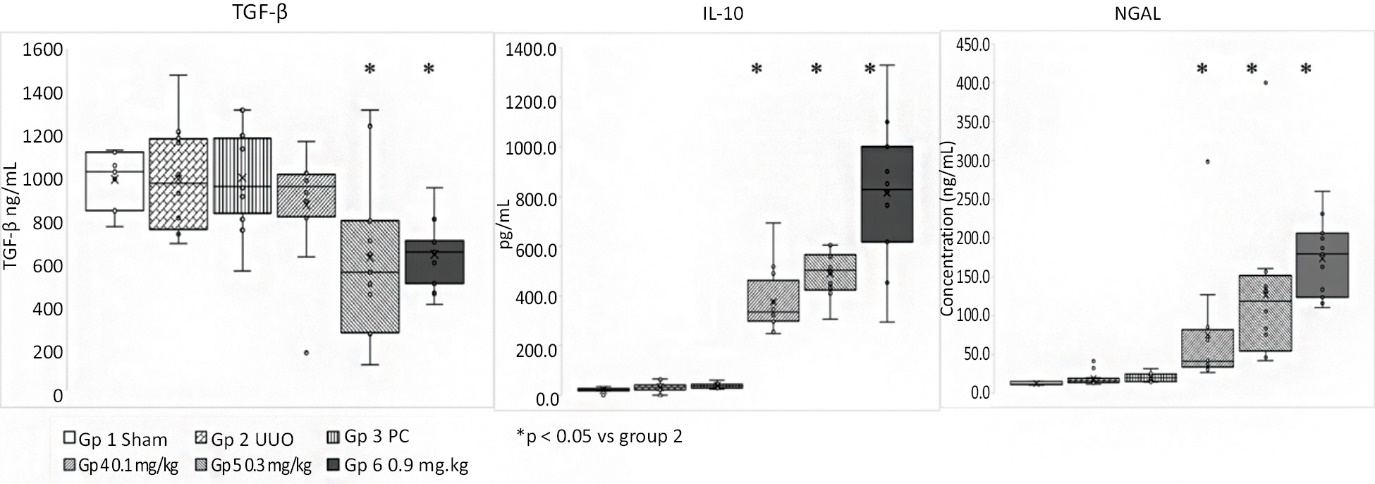
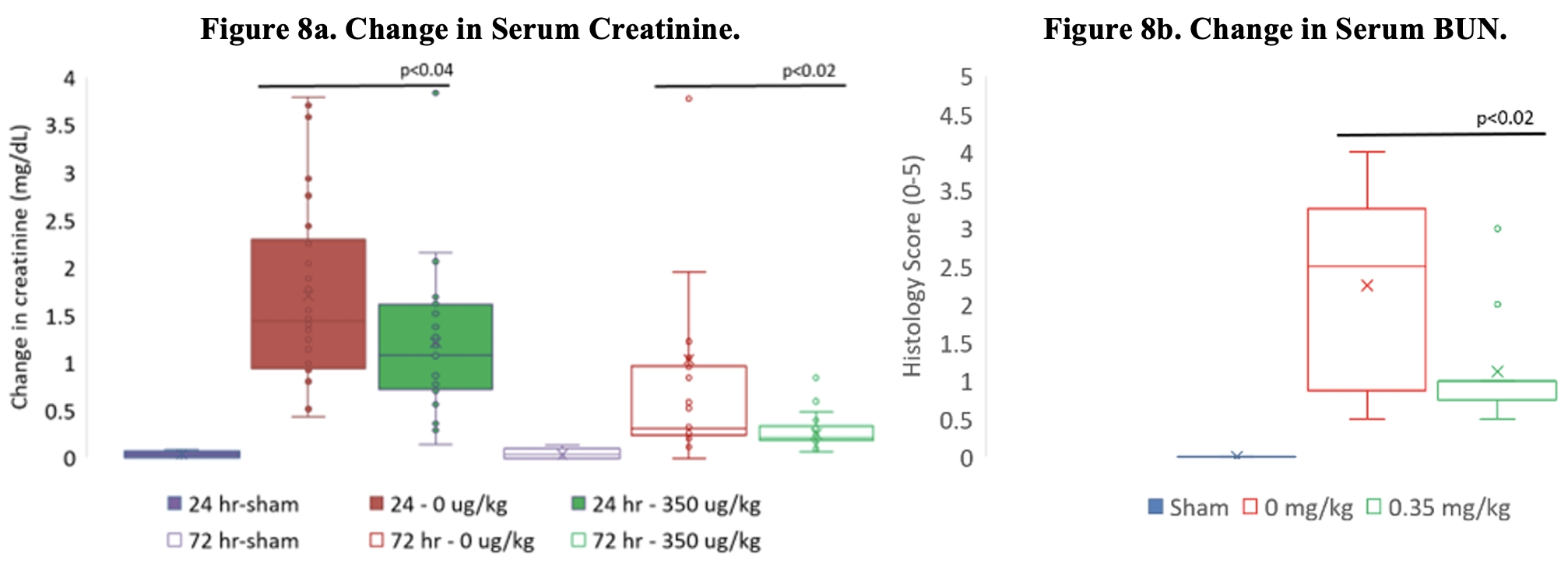
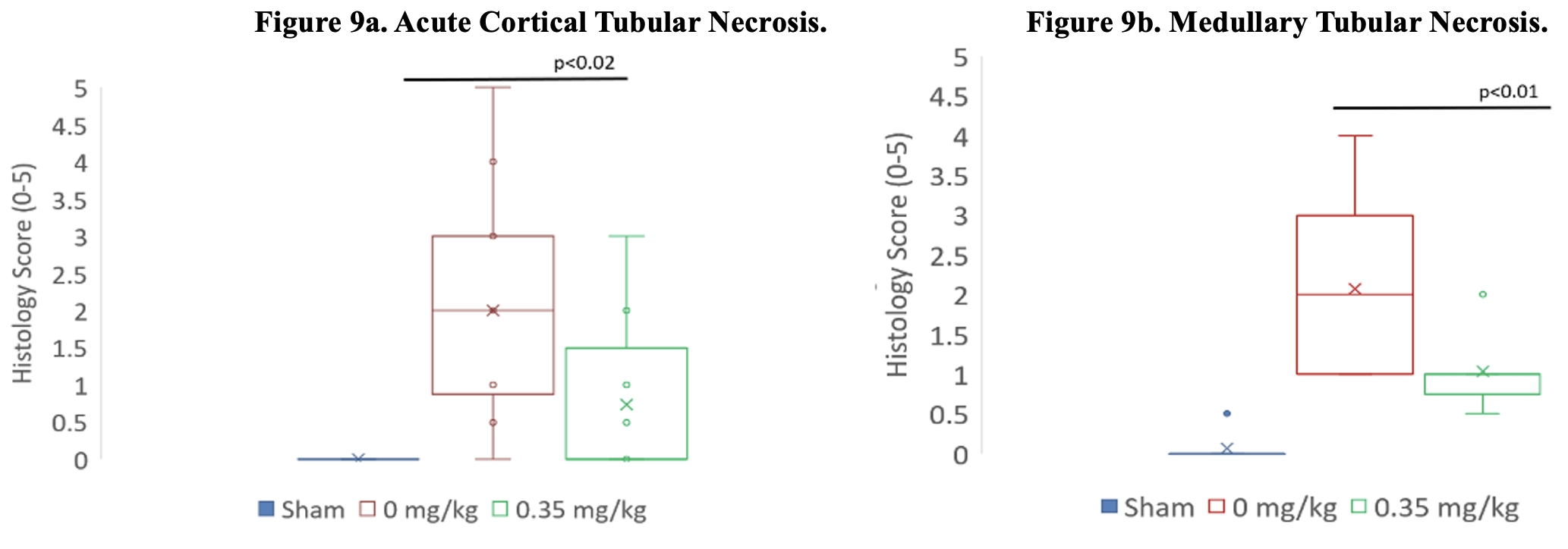
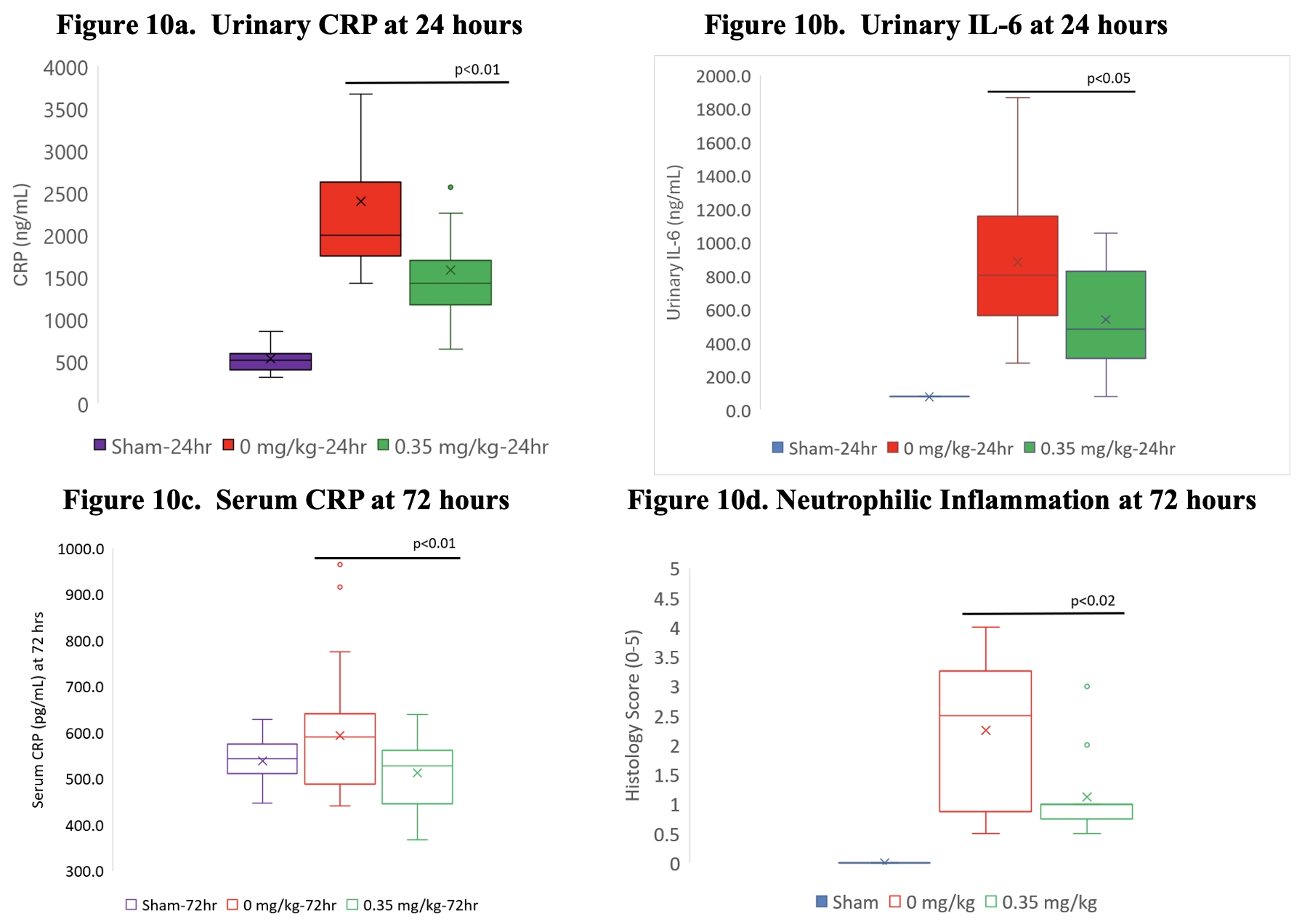
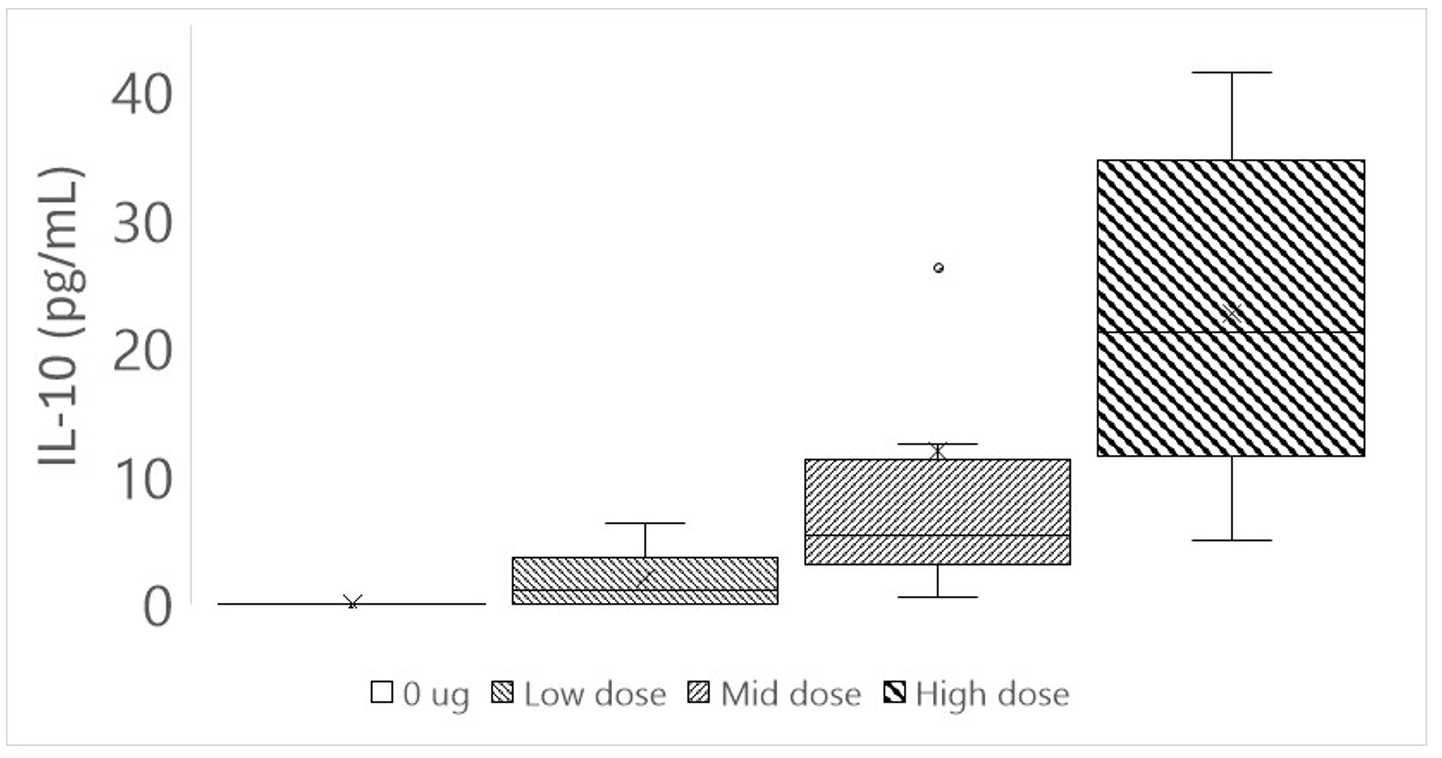
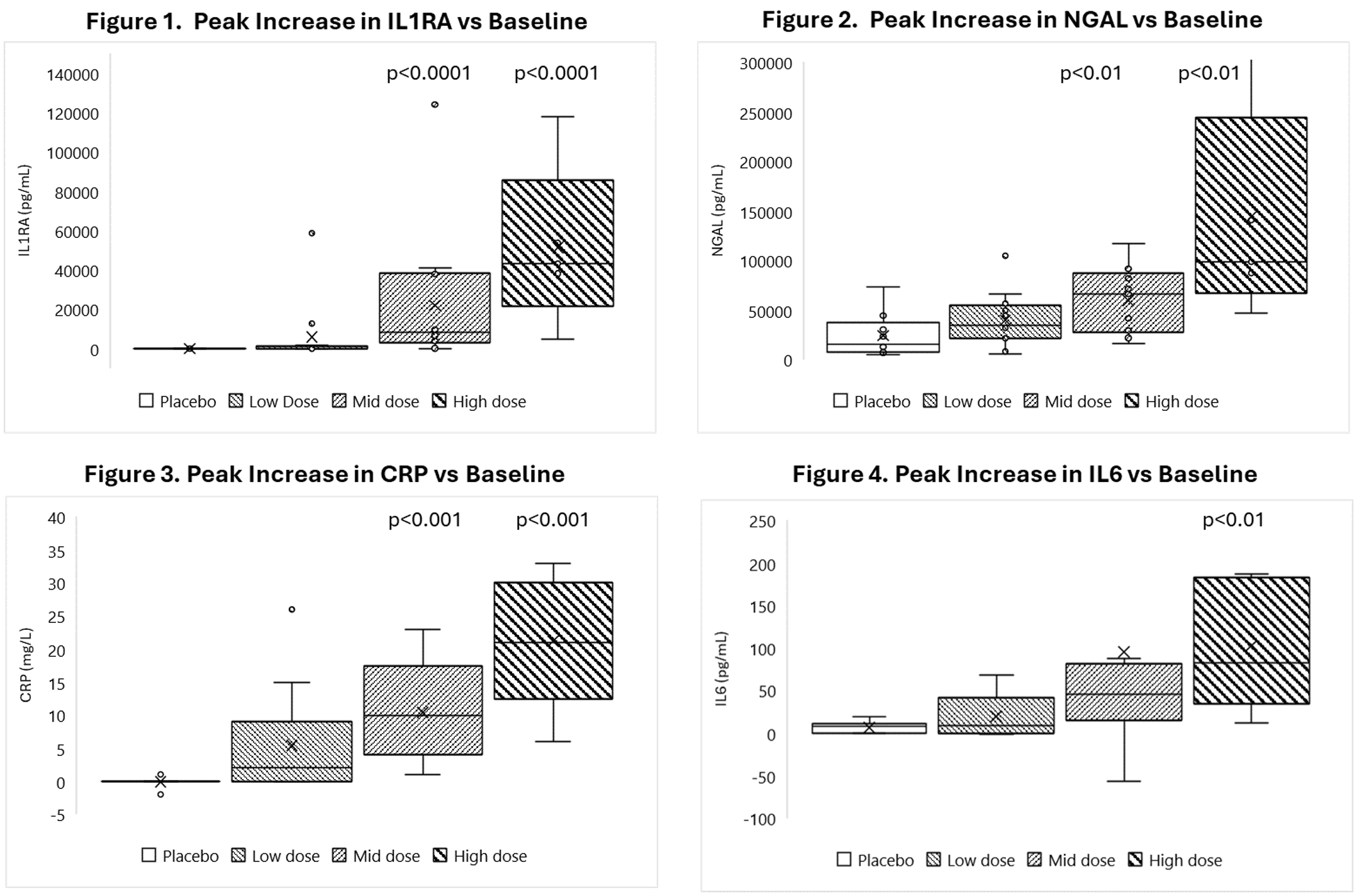
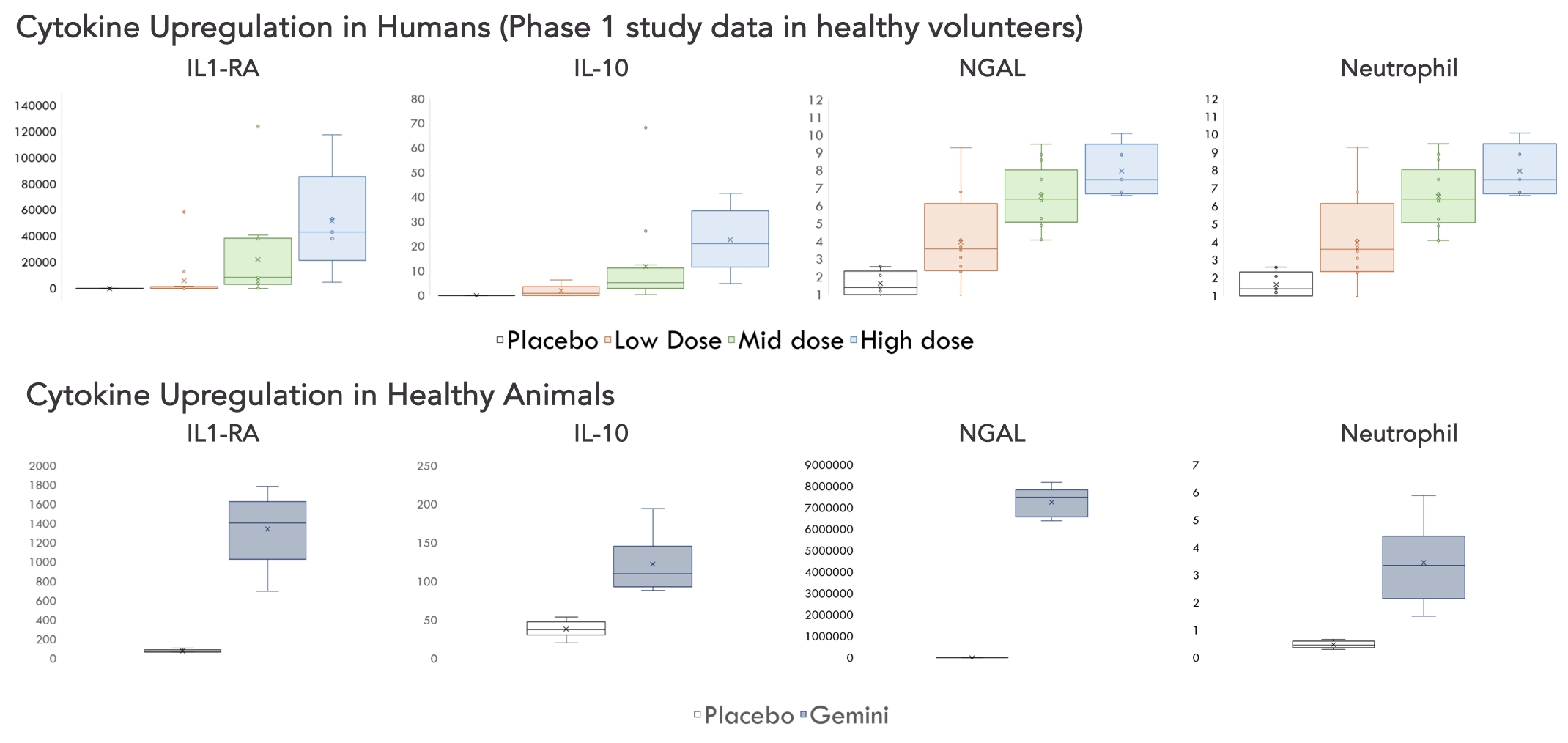
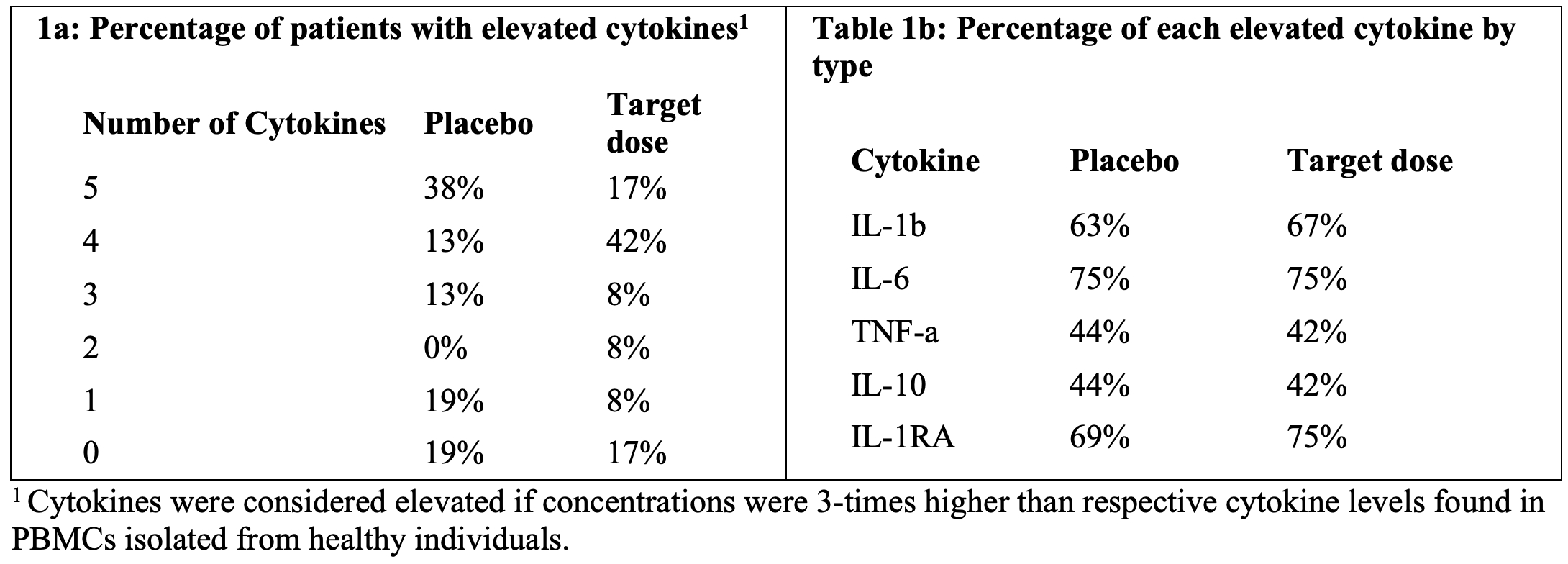
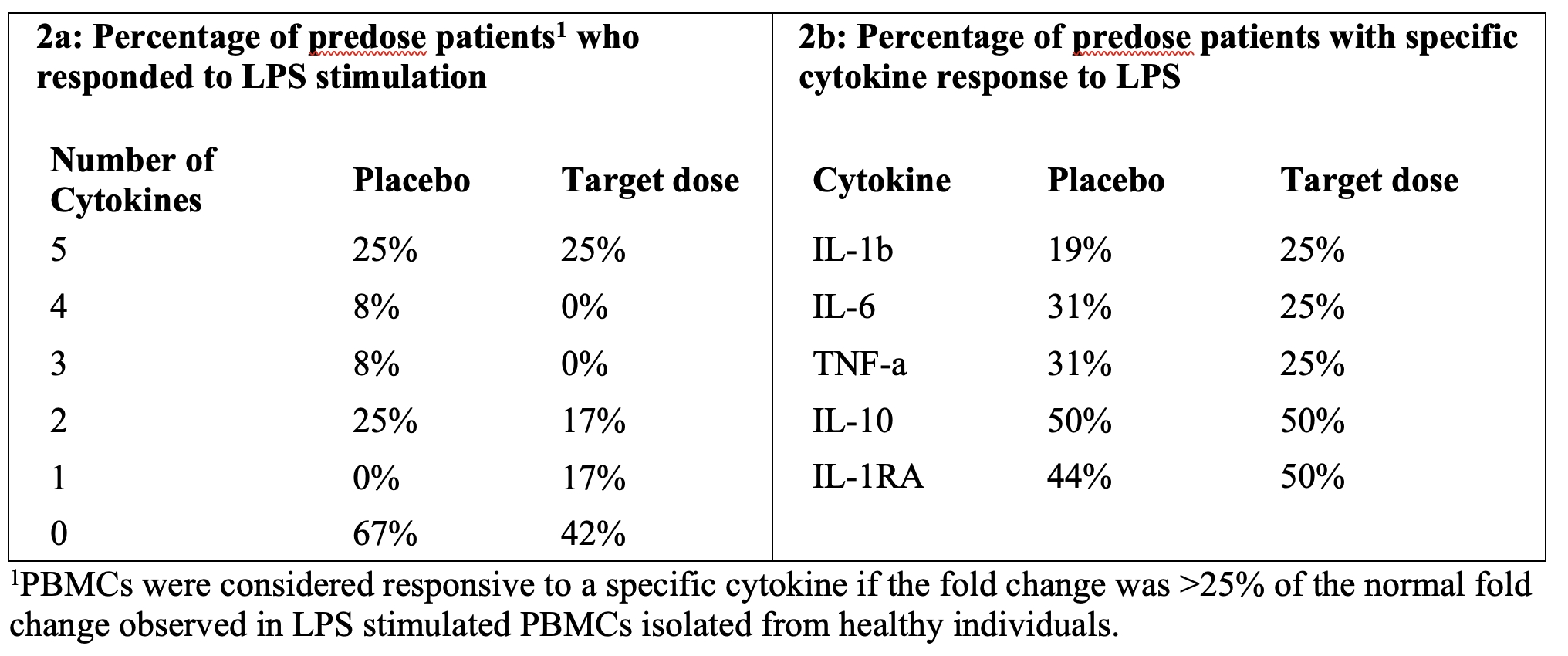
As of February 23, 2026, our patent portfolio includes one patent family directed to MPLA formulations, including Gemini. This patent family includes one U.S. application, one European Patent Organization (“EPO”) application, and one Canadian application. Our portfolio additionally includes a second patent family, consisting of a U.S application and a Canadian application directed to methods of using MPLA formulations, including Gemini, as an adjuvant to traditional allergy immunotherapy, such as oral allergy immunotherapy. Our portfolio also includes a U.S. provisional patent application covering dosing regimens of MPLA formulations. Regarding our GEM-AKI and GEM-CKD programs, our portfolio also includes a patent family directed to the use of MPLA formulations for the prevention of loss of function associated with acute organ disease and chronic organ disease. This patent family includes one application in the U.S., as well as applications filed in the EPO, China, Hong Kong, Japan, South Korea, and Canada. Finally, we have licensed from Vanderbilt University a patent directed to methods of using PHAD for treating or preventing infections.
Our pending and future patent applications may not result in patents being issued which protect our technology or Product Candidates, or which effectively prevent others from commercializing competitive technologies and Product Candidates.
Generally, issued patents are granted a term of 20 years from the earliest claimed non-provisional filing date. In certain instances, patent term can be adjusted to recapture a portion of delay incurred by the U.S. Patent and Trademark Office (the “USPTO”) in examining the patent application (patent term adjustment, or “PTA”) or extended to account for term effectively lost as a result of the FDA regulatory review period (patent term extension, or “PTE”), or both. In addition, we cannot provide any assurance that any patents will be issued from our pending or future applications or that any issued patents will adequately protect our products or Product Candidates.
We believe that we have certain know-how and trade secrets relating to our technology and Product Candidates. We rely on trade secrets to protect certain aspects of our technology related to our current and future product candidates. However, trade secrets can be difficult to protect. We seek to protect our trade secrets, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors, service providers, and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems.
Employees
As of February 23, 2026, we had 8 full-time employees and one part-time employee, 5 of whom are engaged in research and development activities or operations and 4 of whom are engaged in general and administrative activities or operations. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We consider our relationship with our employees to be positive.
Government Regulation
The FDA and other regulatory authorities at federal, state and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, recordkeeping, approval, advertising, promotion, marketing, post-approval monitoring and post-approval reporting of drugs. We, along with our vendors, contract research organizations (“CROs”), clinical investigators and contract manufacturing organizations (“CMOs”) will be required to navigate the various preclinical, clinical, manufacturing and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval of our Product Candidates. The process of obtaining regulatory approvals of drugs and ensuring subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources.
In the United States, the FDA regulates drug products under the Federal Food, Drug, and Cosmetic Act (“FD&C Act”), its implementing regulations, and other federal, state and local statutes and regulations. Drugs are also subject to other federal, state and local statutes and regulations. If we fail to comply with applicable FDA or other requirements at any time with respect to product development, clinical testing, approval or any other regulatory requirements relating to product manufacture, processing, handling, storage, quality control, safety, marketing, advertising, promotion, packaging, labeling, export, import, distribution, or sale, we may become subject to administrative or judicial sanctions or other legal consequences. These sanctions or consequences could include, among other things, the FDA’s refusal to approve pending applications, issuance of clinical holds for ongoing studies, suspension or revocation of approved applications, warning or untitled letters, product withdrawals or recalls, product seizures, relabeling or repackaging, total or partial suspensions of manufacturing or distribution, injunctions, fines, civil penalties or criminal prosecution.
Our Product Candidates must be approved for therapeutic indications by the FDA before they may be marketed in the United States. For Product Candidates regulated under the FD&C Act, FDA must approve a New Drug Application (“NDA”). The process generally involves the following:
•completion of extensive preclinical studies in accordance with applicable regulations, including studies conducted in accordance with good laboratory practice (“GLP”) requirements;