FORM 20-F
☐ REGISTRATION STATEMENT PURSUANT TO SECTION
12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934
OR
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR
15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2024
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13
OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
OR
☐ SHELL COMPANY REPORT PURSUANT TO SECTION
13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Commission File No.: 001-39957
NLS PHARMACEUTICS LTD.
(Exact name of registrant as specified in its charter)
Translation of registrant’s name into
English: Not applicable
Switzerland
(Jurisdiction of incorporation or organization)
The Circle 6
8058 Zurich
Switzerland
(Address of principal executive offices)
Alexander Zwyer
Chief Executive Officer
Tel: +41 44 512 21 50
contact@nls-pharma.com
The Circle 6, Postfach
8058 Zurich
Switzerland
(Name, Telephone, E-mail and/or Facsimile number
and Address of Company Contact Person)
Securities registered or to be registered pursuant
to Section 12(b) of the Act:
| Title of each class | | Trading Symbol(s) | | Name of each exchange on which registered |
| Common shares, nominal value CHF 0.03 per share (CHF 0.8 as of December 31, 2024) | | NLSP | | Nasdaq Capital Market |
| Warrants to purchase common shares, nominal value CHF 0.03 per share | | NLSPW | | Nasdaq Capital Market |
Securities registered or to be registered pursuant
to Section 12(g) of the Act: None
Securities for which there is a reporting obligation
pursuant to Section 15(d) of the Act: None
Indicate the number of outstanding shares of each of the issuer’s
classes of capital or common stock as of the close of the period covered by the annual report: 3,159,535 common shares, nominal value
CHF 0.03 per share, as of December 31, 2024.
Indicate by check mark if
the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐ No ☒
If this report is an annual
or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the
Exchange Act of 1934.
Yes ☐ No ☒
Indicate by check mark whether
the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the preceding 12 months
(or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements
for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether
the registrant has submitted every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding
12 months.
Yes ☒ No ☐
Indicate by check mark whether
the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition
of “large accelerated filer, “accelerated filer,” and emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ | Non-accelerated filer ☒ |
| | | Emerging growth company ☒ |
If an emerging growth company
that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the
extended transition period for complying with any new or revised financial accounting standards † provided pursuant to Section
13(a) of the Exchange Act. ☐
| † | The term “new or revised financial accounting standard”
refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012. |
Indicate by check mark whether
the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control
over financial reporting under Section 404(b) of the Sarbanes Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that
prepared or issued its audit report. ☐
If securities are registered
pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing
reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether
any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the
registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark which
basis of accounting the registrant has used to prepare the financial statements included in this filing.
U.S. GAAP ☒
International Financial Reporting
Standards as issued by the International Accounting Standards Board ☐
Other ☐
If “Other” has
been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to
follow.
☐ Item 17 ☐ Item
18
If this is an annual report, indicate by check
mark whether the registrant is a shell company.
Yes ☐ No ☒
In this annual report, “we,”
“us,” “our,” the “Company” and “NLS” refer to NLS Pharmaceutics Ltd., a Swiss corporation,
and its wholly owned subsidiary, NLS Pharmaceutics Inc., a Delaware corporation. “Kadimastem” refers to Kadimastem Ltd.,
an Israeli publicly traded company limited by shares (TASE: KDST). “Merger Sub” refers to NLS Pharmaceutics (Israel) Ltd.,
an Israeli company and a wholly owned subsidiary of the Company. “Merger Agreement” refers to the Agreement and Plan of Merger
entered into by and between NLS, Merger Sub and Kadimastem, pursuant to which Merger Sub will merge with and into Kadimastem, with Kadimastem
as the surviving company, or the “Merger.”
Unless the context otherwise
indicates or requires, the NLS logo and all product names and trade names used by us in this annual report, including Quilience®
and Nolazol®, are our proprietary trademarks and service marks. Quilience® and Nolazol® are NOT approved drugs. They
refer to Mazindol ER an investigational drug currently in late stage clinical development by NLS. All trademarks or trade names referred
to in this annual report are the property of their respective owners. Solely for convenience, the trademarks and trade names in this annual
report are referred to without the ® and ™ symbols, but such references should not be construed as any indicator
that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend the use
or display of other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by,
any other companies.
Our reporting currency and
functional currency is the United States, or U.S., dollar. Unless otherwise expressly stated or the context otherwise requires, references
in this annual report to “dollars,” “USD” or “$” mean U.S. dollars, and references to “CHF”
are to the Swiss Franc. U.S. dollar translations of CHF amounts presented in this annual report were done on different dates in accordance
with the date as of such entry in the Company’s books and are derived from our audited financial statements included elsewhere in
this annual report. U.S. dollar translations of CHF amounts presented in this annual report that are not derived from our audited financial
statements included elsewhere in this annual report are translated using the rate of CHF 1.00 to $1.0965, based on the exchange rate provided
by the Swiss Federal Tax Administration on December 31, 2024.
This annual report includes
statistical, market and industry data and forecasts which we obtained from publicly available information and independent industry publications
and reports that we believe to be reliable sources. These publicly available industry publications and reports generally state that they
obtain their information from sources that they believe to be reliable, but they do not guarantee the accuracy or completeness of the
information. Although we believe that these sources are reliable, we have not independently verified the information contained in such
publications. Further, Kadimastem has supplied all information contained or incorporated by reference into this annual report relating
to Kadimastem.
On September 25, 2024,
we effectuated a 1-for-40 reverse share split in order to regain compliance with Nasdaq Listing Rule 5550(a)(2). All historical
quantities of Common Shares and per share data herein are presented on a post-split basis to give effect to our 1-for-40 reverse
share split effected prior to the start of trading on the Nasdaq Capital Market, or Nasdaq, on September 27, 2024. Further, on
January 14, 2025, the shareholders of the Company approved a change in the par value of our Common Shares from CHF 0.80 to CHF 0.03
per share, effective January 17, 2025. All share amounts reflect the par value of CHF 0.80 as of December 31, 2024 and no
adjustments have been made for the change in par value to additional paid in capital for
the reduction in par value.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Certain information included
in this annual report on Form 20-F, or this annual report, may be deemed to be “forward-looking statements,” including some
of the statements made under Item 3.D. “Risk Factors,” Item 5 “Operating and Financial Review and Prospects,”
“Business” and elsewhere in this annual report constitute forward-looking statements. Forward-looking statements are often
characterized by the use of forward-looking terminology such as “may,” “will,” “expect,” “anticipate,”
“estimate,” “continue,” “believe,” “predict,” “should,” “intend,”
“project” or other similar words, but are not the only way these statements are identified.
These forward-looking statements
may include, but are not limited to, statements relating to our objectives, plans and strategies, statements that contain projections
of results of operations or of financial condition, expected capital needs and expenses, statements relating to the research, development,
completion and use of our product candidates, and all statements (other than statements of historical facts) that address activities,
events or developments that we intend, expect, project, believe or anticipate will or may occur in the future.
Forward-looking statements
are not guarantees of future performance and are subject to risks and uncertainties. We have based these forward-looking statements on
assumptions and assessments made by our management in light of their experience and their perception of historical trends, current conditions,
expected future developments and other factors they believe to be appropriate.
Important factors that could
cause actual results, developments and business decisions to differ materially from those anticipated in these forward-looking statements
include, among other things:
| |
● |
the regulatory pathways that we may elect to utilize in seeking European Medicines Agency, or EMA, the U.S. Food and Drug Administration, or FDA, and other regulatory approvals; |
| |
|
|
| |
● |
the completion and timing of the transaction contemplated by our Merger Agreement with Kadimastem are uncertain, and the risk that the transaction may not close as expected or at all; |
| |
|
|
| |
● |
our ability to drive revenue growth, enhance research and development capabilities, and improve financial performance as a result of the potential merger with Kadimastem is subject to uncertainties, including unforeseen costs and integration issues; |
| |
|
|
| |
● |
that our financial position raises substantial doubt about our ability to continue as a going concern; |
| |
|
|
| |
● |
our ability to maintain listing and effectively comply with the listing requirements of the Nasdaq; |
| |
● |
the re-scheduling of Mazindol ER in the U.S. by the U.S. Drug Enforcement Agency, or the DEA, following approval by the FDA; |
| |
● |
the launch of a different formulation or different dosage of Mazindol by another company; |
| |
● |
the use of Quilience (Mazindol ER) in a compassionate use program, or CUP, and the results thereof; |
| |
● |
obtaining EMA and FDAFDA approval of, or other regulatory action in Europe or the United States and elsewhere with respect to, Quilience, Nolazol, NLS-4, or other product candidates that we may seek to develop; |
| |
● |
the commercial launch and future sales of Quilience and/or Nolazol, or any other future product candidates; |
| |
● |
the dosage of Quilience, Nolazol, and or any of our pipeline drugs; |
| |
|
|
| |
● |
our ability to move NLS-3, NLS-8, NLS-11, NLS-12 and any of our Aexon Labs (Dual) Orexin compounds (recently secured through an in-license) into investigational new drug, or IND-enabling studies; |
| |
|
|
| |
● |
our expectations regarding the timing of commencing further clinical trials, the process entailed in conducting each such trial, including dosages, and the order of such trials with each of our product candidates or whether such trials will be conducted at all; |
| |
● |
improved convenience relating to the prescription of and use of Nolazol for prescribers and patients (and their parents); |
| |
● |
our expectations regarding the supply of mazindol; |
| |
● |
third-party payor reimbursement for Quilience, Nolazol, and or any of our pipeline drugs; |
| |
|
|
| |
● |
our estimates regarding anticipated expenses, capital requirements and our needs for additional financing; |
| |
● |
changes to the narcolepsy patient market size and market adoption of Quilience by physicians and patients; |
| |
● |
the timing, cost, regulatory approvals or other aspects of the commercial launch of Quilience and Nolazol; |
| |
● |
submission of a Marketing Authorisation Application, or MAA, and New Drug Application, or NDA, with the EMA and FDA for Quilience, Nolazol, and or any of our pipeline drugs, respectively; |
| |
● |
completion and receiving favorable results of clinical trials for Quilience, Nolazol, and or any of our pipeline drugs; |
| |
|
|
| |
● |
issuance of patents to us by the U.S. Patent and Trademark Office, or PTO, and other governmental patent agencies; |
| |
● |
new issuances of orphan drug designations; |
| |
● |
the overall global political and economic environment in the countries in which we operate; |
| |
|
|
| |
● |
the development and approval of the use of mazindol for additional indications other than narcolepsy and attention deficit hyperactivity disorder, or ADHD; |
| |
|
|
| |
● |
the development and commercialization, if any, of any other product candidates that we may seek to develop; |
| |
|
|
| |
● |
the use of mazindol controlled release, or CR, for treatment of additional indications other than narcolepsy, idiopathic hypersomnia, or IH, and ADHD; and |
| |
● |
the ability of our management team to lead the development of our product candidates, conclude a strategic partnership deal for Mazindol or any of our pipeline compounds. |
These statements are only
current predictions and are subject to known and unknown risks, uncertainties and other factors that may cause our or our industry’s
actual results, levels of activity, performance or achievements to be materially different from those anticipated by the forward-looking
statements. We discuss many of these risks in this in greater detail under Item 3.D. “Risk Factors” and elsewhere in this
annual report. You should not rely upon forward-looking statements as predictions of future events. Readers are urged to carefully review
and consider the various disclosures made throughout this annual report which are designed to advise interested parties of the risks and
factors that may affect our business, financial condition, results of operations and prospects.
You should not put undue reliance
on any forward-looking statements. Any forward-looking statements in this annual report are made as of the date hereof, and we undertake
no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise,
except as required by law.
In addition, the section of
this annual report entitled “Item 4. Information on the Company” contains information obtained from independent industry sources
and other sources that we have cited but not independently verified.
PART I
ITEM 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT
AND ADVISERS
Not applicable.
ITEM 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not applicable.
ITEM 3. KEY INFORMATION
A. [Reserved.]
B. Capitalization and Indebtedness.
Not applicable.
C. Reasons for the Offer and Use of Proceeds.
Not applicable.
D. Risk Factors.
You should carefully consider
the risks described below, together with all of the other information in this annual report. The risks described below are not the only
risks facing us. Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial may also materially
and adversely affect our business operations. If any of these risks actually occurs, our business and financial condition could suffer
and the price of our common shares and our publicly listed warrants, or Warrants, could decline.
Summary of Risk Factors
Investing in our common shares
and Warrants involves substantial risks. Our ability to execute our strategy is also subject to certain risks. The risks described below
may cause us not to realize the full benefits of our strengths or may cause us to be unable to successfully execute all or part of our
strategy. In particular, our risks include, but are not limited to, the following summary of such risk factors:
| |
● |
We may be unable to successfully use mazindol, on which we depend substantially, as the drug substance for each of our current clinical mid-stage product candidates, Quilience, for the treatment of narcolepsy, and Nolazol, for the treatment of ADHD, and which outcome could prove costly to our business and could prevent us from obtaining regulatory or marketing approval; |
| |
|
|
| |
● |
We may not be able to initiate our Phase 3 clinical trials in Quilience without additional pre-clinical studies, chemistry, manufacturing, and controls, or CMC, work or early-stage clinical trials; |
| |
● |
Prior results of mazindol for the treatment of other indications may not be replicated in the clinical trials that we conduct for the treatment of narcolepsy or ADHD; |
| |
|
|
| |
● |
If, and when, we seek to commercialize Quilience and/or Nolazol, we may be partially dependent upon prescriptions from physicians for the sale of both such product candidates, and therefore, the loss of a significant number of patient referrals by physicians prescribing Quilience and/or Nolazol may have an adverse effect on our future revenues, if any, which could have an adverse effect on our business, financial condition and results of operations; |
| |
● |
We are dependent on a sole manufacturer for mazindol drug substance and product. Any delay, price increase, or unavailability could materially disrupt our clinical development or commercialization efforts; |
| |
● |
Failure to secure additional strategic partnerships could negatively affect our commercialization prospects; |
| |
● |
If we are unable to obtain and maintain effective patent rights for our products, we may not be able to compete effectively in our markets. If we are unable to protect the confidentiality of our trade secrets or know-how, such proprietary information may be used to compete against us; |
| |
● |
If we are unable to maintain effective proprietary rights for our products, we may not be able to compete effectively in our markets; |
| |
● |
We may face future claims that our personnel improperly used or disclosed third-party confidential information or trade secrets from prior employers; |
| |
● |
Our financial statements for the year ended December 31, 2024, contained a going concern disclosure in Note 1 regarding substantial doubt about our ability to continue as a going concern. This going concern disclosure in Note 1 of financial statements could prevent us from obtaining new financing on reasonable terms or at all and risk our ability to continue operating as a going concern; |
| |
● |
To date, we have not generated any revenue, and we do not expect to
generate any significant revenue unless and until we obtain marketing approval to commercialize Quilience and/or Nolazol. We are unable
to predict the extent of future losses or when we will become profitable based on the sale of any product, if at all. Even if we succeed
in developing and commercializing our product candidates, we may never generate sufficient revenue to sustain profitability. As of December
31, 2024, we had an accumulated deficit of approximately $74.4 million; |
| |
● |
We have not generated revenue, have a history of losses, and expect to continue incurring losses. We will need substantial additional capital to advance our pipeline, including Quilience, Nolazol, other candidates (NLS-3, NLS-4, NLS-8, NLS-11, NLS-12), and the recently in-licensed Aexon platform, which may not be available or may only be available on unfavorable terms; |
| |
|
|
| |
● |
As a public company, we must comply with extensive U.S., Swiss, and Nasdaq regulations, which are costly and require significant management attention. We may be delisted from Nasdaq due to non-compliance or financial constraints; |
| |
● |
We require regulatory approvals, including from the FDA and EMA, to commercialize our products; |
| |
● |
Clinical data may not satisfy regulatory authorities or may require additional studies, delaying or preventing approval; |
| |
● |
As a foreign private issuer and an emerging growth company, we follow reduced U.S. reporting standards, which could impact investor confidence; |
| |
|
|
| |
● |
We have identified material weaknesses in our internal controls; failure to remediate them may impair financial reporting and harm investor trust; |
| |
● |
If the Merger with Kadimastem is not consummated, NLS’s share price and business could be adversely affected; |
| |
|
|
| |
● |
Following the Merger, NLS may lose its foreign private issuer status and face increased U.S. reporting and compliance obligations; |
| |
● |
The Merger with Kadimastem may result in dilution and reduced influence for current NLS shareholders. |
| |
● |
NLS may not be able to successfully integrate Kadimastem’s business or realize the expected benefits of the Merger; |
| |
● |
Following the Merger, NLS plans to shift its business to Kadimastem’s cell therapy platform, which may not succeed; |
| |
● |
Kadimastem has no revenue and a history of losses, and its drug candidates may never receive regulatory approval or be commercialized; and |
| |
● |
Legal or regulatory challenges, including lawsuits, could delay or prevent the completion of the Merger with Kadimastem. |
Risks Related to Our Current Business
We depend substantially on the success of
our two product candidates, Quilience and Nolazol. We cannot give any assurance that any of our product candidates will receive regulatory
approval, which is necessary before they can be commercialized.
We have invested
almost all of our efforts and financial resources to achieve and maintain phase 3 readiness in research and development and general and
administrative costs of our two product candidates, Quilience for the treatment of excessive daytime sleepiness, or EDS, and cataplexy
associated with narcolepsy and Nolazol, for the treatment of ADHD. The process to develop, obtain regulatory approval for and commercialize
pharmaceutical product candidates is long, complex, costly and inherently uncertain of outcome. We are not permitted to market any of
our product candidates in the United States, European Union, or the EU, or any other jurisdiction until we receive the requisite regulatory
approvals. We may be able to generate pre-approval revenues from compassionate use activities leveraging on an expanded access policy
in certain countries around the world. However, we cannot give any assurance that our current clinical development plan will proceed as
planned, or that our product candidates will receive regulatory approval, or that such regulatory approval, if received, will be within
a timeframe that allows us to effectively compete with our competitors, or be successfully marketed and commercialized.
Our initiation of clinical trials for Quilience
is dependent upon the review and approval of the relevant regulatory agencies and authorities and if we are required to conduct pre-clinical
trials, approval of an NDA or MAA, if any, for Quilience would be delayed and we may require additional capital as a result thereof.
Based in part on the prior
use and FDA approval of mazindol to manage exogenous obesity, we have been able to commence our Phase 2 clinical trials for Quilience
without having to do prior pre-clinical and/or early-stage clinical trials, such as Phase 1 trials. No assurance can be given that the
EMA or FDA will agree to allow us to initiate Phase 3 clinical trials for Quilience without conducting such pre-clinical clinical trials.
If the FDA or EMA, or any other applicable regulatory agency, were to require us to conduct additional pre-clinical trials, our planned
development strategy for Quilience would be materially impacted and approval of an NDA or MAA, if any, for Quilience would be delayed
and we may require additional capital as a result thereof.
In addition, we may request
a Paediatric Investigation Plan, or PIP, deferral in order to delay conducting clinical trials for Quilience in children until after we
receive an MAA from the EMA for the use of Quilience in adults. A PIP deferral, as can be agreed upon by the EMA, allows an applicant
to delay studies in children until after there is sufficient data on use in adults; we anticipate that we will be able to receive a PIP
deferral from the EMA for Quilience; however, there is no guarantee that this deferral will be granted, which could impact our planned
development process and would make the product candidate approval process more costly.
The commencement and completion of clinical
trials can be delayed or prevented for a number of reasons.
We may not be able to commence
or complete the clinical trials that would support our submission of an NDA to the FDA or MAA to the EMA. Drug development is a long,
expensive and uncertain process, and delay or failure can occur at any stage of any of our clinical trials. Clinical trials can be delayed
or prevented for a number of reasons, including:
| |
● |
difficulties obtaining regulatory approval to commence a clinical trial or complying with conditions imposed by a regulatory authority regarding the scope or term of a clinical trial; |
| |
|
|
| |
● |
delays in reaching or failing to reach agreement on acceptable terms with prospective contract research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| |
● |
insufficient or inadequate supply or quality of a product candidate or other materials necessary to conduct our clinical trials; |
| |
● |
if the FDA or EMA elect to enact policy changes, as a result of a pandemic threat or otherwise; |
| |
● |
difficulties obtaining institutional review board, or IRB, approval to conduct a clinical trial at a prospective site; and |
| |
|
|
| |
● |
challenges recruiting and enrolling patients to participate in clinical trials for a variety of reasons, including size and nature of patient population, proximity of patients to clinical sites, eligibility criteria for the trial, nature of trial protocol, the availability of approved effective treatments for the relevant disease and competition from other clinical trial programs for similar indications. |
Clinical trials may also be
delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated
by us, the FDA, the IRBs at the sites where the IRBs are overseeing a trial, a data safety monitoring board overseeing the clinical trial
at issue or by other regulatory authorities due to a number of factors, including:
| |
● |
failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| |
● |
inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities; |
| |
● |
unforeseen safety issues (including those that result from COVID-19) or lack of effectiveness; and |
| |
● |
lack of adequate funding to continue the clinical trial. |
The results of pre-clinical studies, early-stage
clinical trials, data obtained from real-world use, and published third-party studies may not be indicative of results in future clinical
trials and we cannot assure you that any planned or future clinical trials will lead to results sufficient for the necessary regulatory
approvals.
The results of pre-clinical
studies may not be predictive of the results of clinical trials, and the results of any completed clinical trials, including studies derived
from real-world use and studies in published literature, or clinical trials we commence may not be predictive of the results of later-stage
clinical trials. Additionally, interim results during a clinical trial do not necessarily predict final results. Later clinical trial
results may not replicate earlier clinical trials for a variety of reasons, including differences in trial design, different trial endpoints
(or lack of trial endpoints in exploratory studies), subject population, number of subjects, subject selection criteria, trial duration,
drug dosage and formulation and lack of statistical power in the earlier studies. There can be no assurance that any of our clinical trials
will ultimately be successful or support further clinical development of any of our product candidates. A number of companies in the pharmaceutical
and biotechnology industries have suffered significant setbacks in clinical development even after achieving promising results in earlier
studies, and any such setbacks in our clinical development could have a negative impact on our business.
We may find it difficult to enroll patients
in our clinical trials. Difficulty in enrolling patients could delay or prevent clinical trials of our product candidates.
Identifying and qualifying
patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends
in part on the speed at which we can recruit patients to participate in testing our product candidates, and we may experience delays in
our clinical trials if we encounter difficulties in enrollment.
In addition, as a rare disorder,
there is a limited patient pool from which to draw for our clinical trials for Quilience. Further, the eligibility criteria of our clinical
trials will further limit the pool of available study participants as we will require that patients have specific characteristics that
we can measure or to assure their disease is either severe enough or not too advanced to include them in a study.
Additionally, the process
of finding patients may prove costly. We also may not be able to identify, recruit and enroll a sufficient number of patients to complete
our clinical trials because of the perceived risks and benefits of the product candidate under study, the availability and efficacy of
competing therapies and clinical trials, the proximity and availability of clinical trial sites for prospective patients and the patient
referral practices of physicians. If patients are unwilling to participate in our studies for any reason, the timeline for recruiting
patients, conducting studies and obtaining regulatory approval of our potential product candidates will be delayed.
If we experience delays in
the completion or termination of any clinical trial of our product candidates, the commercial prospects of our product candidates will
be harmed, and our ability to generate product candidate revenue from any of these product candidates could be delayed or prevented. In
addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval
process and jeopardize our ability to commence product candidate sales and generate revenues. Any of these occurrences may harm our business,
financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement
or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
Interim topline and preliminary data from
our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to
audit and verification procedures that could result in material changes in the final data.
From time to time, we may
publish interim topline or preliminary data from our clinical trials. Interim data from clinical trials that we may complete are subject
to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become
available. Preliminary or topline data also remain subject to audit and verification procedures that may result in the final data being
materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with
caution until the final data are available. Adverse differences between preliminary or interim data and final data could significantly
harm our reputation and business prospects.
Changes in methods of product candidate
manufacturing or formulation may result in additional costs or delay.
As product candidates proceed
through pre-clinical studies to late-stage clinical trials towards potential approval and commercialization, it is common that various
aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an effort to optimize
processes and results. Such changes carry the risk that they will not achieve these intended objectives. Any of these changes could cause
our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted
with the materials manufactured using altered processes. Such changes may also require additional testing, FDA or EMA notification or
FDA approval. This could delay completion of clinical trials, require the conduct of bridging clinical trials or the repetition of one
or more clinical trials, increase clinical trial costs, delay approval of our product candidates and jeopardize our ability to commence
sales and generate revenues.
International expansion of our business
exposes us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the
United States, Switzerland or the EU.
Other than our
headquarters and other operations which are located in Switzerland and our wholly owned U.S. subsidiary, NLS Pharmaceutics Inc., a
Delaware corporation (as further described below), we currently have limited international operations, but our business strategy
incorporates potentially significant international expansion, particularly in anticipation of approval of our product candidates. We
may plan to maintain sales representatives and conduct physician and patient association outreach activities, as well as clinical
trials, outside of the United States, Switzerland and Europe. If Quilience and/or Nolazol or any of our other product candidates are
approved for commercialization outside the United States, Switzerland, or the EU, we will likely enter into agreements with third
parties to market the drugs in these additional global territories. We expect that we will be subject to additional risks related to
entering into or maintaining international business relationships, including:
| |
● |
different regulatory requirements for drug approvals in foreign countries; |
| |
● |
differing United States and foreign drug import and export rules, tariffs and other trade barriers; |
| |
● |
reduced protection for intellectual property rights in foreign countries; |
| |
● |
failure by us to obtain regulatory approvals for the use of our products in various countries; |
| |
● |
different reimbursement systems; |
| |
● |
economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| |
● |
multiple, conflicting and changing laws and regulations such as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses; |
| |
● |
complexities associated with managing multiple payor reimbursement regimes, government payors or patient self-pay systems; |
| |
● |
financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for our products and exposure to foreign currency exchange rate fluctuations, which could result in increased operating expenses and reduced revenues; |
| |
● |
workforce uncertainty in countries where labor unrest is more common than in the United States; |
| |
● |
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; |
| |
|
|
| |
● |
regulatory and compliance risks that relate to maintaining accurate information and control over sales and activities that may fall within the purview of the U.S. Foreign Corrupt Practices Act, or FCPA, its books and records provisions or its anti-bribery provisions; |
| |
● |
business interruptions resulting from cyber-attacks, geo-political actions, including war (such as the Russia-Ukraine war and Israel’s multi-front war) and terrorism, or natural disasters including earthquakes, typhoons, floods and fires. |
| |
● |
potential liability resulting from development work conducted by these distributors; and |
| |
● |
business interruptions resulting from a local or worldwide pandemic, such as COVID-19, geopolitical actions, including war and terrorism, or natural disasters. |
Any of these factors could
significantly harm our future international expansion and operations and, consequently, our results of operations.
Even if any of our product candidates receives
marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the
medical community necessary for commercial success.
The commercial success of
Quilience and/or Nolazol will depend upon the acceptance of each product by the medical community, including physicians, patients and
third-party payors. The degree of market acceptance of any approved product will depend on a number of factors, including:
| |
● |
the efficacy and safety of the product; |
| |
● |
the potential advantages of the product compared to available therapies; |
| |
|
|
| |
● |
the convenience and ease of administration compared to alternative treatments; |
| |
|
|
| |
● |
limitations or warnings, including use restrictions contained in the product’s approved labeling; |
| |
● |
distribution and use restrictions imposed by the EMA, FDA, or other regulatory authority or agreed to by us as part of a mandatory or voluntary risk management plan; |
| |
● |
availability of alternative treatments, including, in the case of Nolazol, a number of competitive products already approved for the treatment of ADHD or expected to be commercially launched in the near future; |
| |
● |
pricing and cost effectiveness in relation to alternative treatments; |
| |
● |
if the product is included under physician treatment guidelines as a first-, second-, or third-line therapy; |
| |
● |
the strength of sales, marketing and distribution support; |
| |
|
|
| |
● |
the availability of third-party coverage and adequate reimbursement and the willingness of patients to pay out-of-pocket in the absence of coverage by third-party payors; |
| |
● |
the strength of sales, marketing and distribution support; |
| |
● |
the willingness of patients to pay for drugs out of pocket in the absence of third-party coverage; and |
| |
|
|
| |
● |
the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies. |
If Quilience and/or Nolazol
is approved but does not achieve an adequate level of acceptance by physicians, third party payors and patients, we may not generate sufficient
revenue from the product, and we may not become or remain profitable. In addition, our efforts to educate the medical community and third-party
payors on the benefits of the product may require significant resources and may never be successful.
In addition, we may choose
to collaborate with third parties that have direct sales forces and established distribution systems, either to augment our own sales
force and distribution systems or in lieu of our own sales force and distribution systems. If we enter into arrangements with third parties
to perform sales, marketing and distribution services for our products, the resulting revenues or the profitability from these revenues
to us are likely to be lower than if we had sold, marketed and distributed our products ourselves. If we are unable to enter into such
arrangements on acceptable terms or at all, we may not be able to successfully commercialize any of our product candidates that receive
regulatory approval. Depending on the nature of the third-party relationship, we may have little control over such third parties, and
any of these third parties may fail to devote the necessary resources and attention to sell, market and distribute our products effectively.
If we are not successful in commercializing our product candidates, either on our own or through collaborations with one or more third
parties, our future product revenue will suffer and we may incur significant additional losses.
Even if we are able to commercialize any
product candidates, the products may become subject to unfavorable pricing regulations or third-party coverage and reimbursement policies,
any of which could harm our business.
Our ability to commercialize
any product candidates successfully will depend, in part, on the extent to which coverage and reimbursement for these products and related
treatments will be available from government health administration authorities, private health insurers and other organizations. Government
authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they
will pay for and impact reimbursement levels.
Obtaining and maintaining
adequate reimbursement for our products may be difficult. We cannot be certain if and when we will obtain an adequate level of reimbursement
for our products by third party payors. Even if we do obtain adequate levels of reimbursement, third-party payors, such as government
or private healthcare insurers, may carefully review and increasingly question the coverage of, and challenge the prices charged for,
our drugs. Reimbursement rates from private health insurance companies vary depending on the company, the insurance plan and other factors.
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have
attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party
payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged
for drugs. We may also be required to conduct expensive pharmacoeconomic studies to justify coverage and reimbursement or the level of
reimbursement relative to other therapies. If coverage and reimbursement are not available or reimbursement is available only to limited
levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval, and the royalties
resulting from the sales of those products may also be adversely impacted.
There may be significant delays
in obtaining reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved
by the FDA or similar regulatory authorities outside the United States. Moreover, eligibility for reimbursement does not imply that a
drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution.
Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent.
Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement
levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be
reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of
laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Our inability
to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products
that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products
and our overall financial condition.
The regulations that govern
marketing approvals, pricing, coverage and reimbursement for new drug products vary widely from country to country. Current and future
legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining
approvals. Some countries require approval of the sale price of a drug before it can be reimbursed. In many countries, the pricing review
period begins after marketing or product licensing approval is granted. In some foreign markets, prescription drug pricing remains subject
to continuing governmental control, including possible price reductions, even after initial approval is granted. As a result, we might
obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch
of the product, possibly for lengthy time periods, and negatively impact the revenues we are able to generate from the sale of the product
in that country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if
our product candidates obtain marketing approval. There can be no assurance that our product candidates, if they are approved for sale
in the United States or in other countries, will be considered medically necessary or cost-effective for a specific indication, or that
coverage or an adequate level of reimbursement will be available.
Our product candidates Quilience and Nolazol
contain the active ingredient mazindol, which is currently listed as a Schedule IV controlled substance under the Controlled Substances
Act of 1970, or CSA. Failure to maintain compliance with applicable requirements under the CSA or a change in the drug enforcement agency,
or the DEA, scheduling (e.g., from IV to III) post approval or other applicable federal or state regulations could have an adverse effect
on our operations and business.
Controlled substances are
classified by the DEA as Schedule I, II, III, IV or V, or CI, CII, CIII, CIV, and CV, substances, with CI substances considered to present
the highest risk of substance abuse and Schedule V substances the lowest risk. The active ingredient of our lead and follow-on product
candidates is mazindol, which is classified as a Schedule IV controlled substance under the CSA, and regulations of the DEA. Under the
CSA, subject to certain exemptions, every person who manufactures, distributes, dispenses, imports or exports any controlled substance
must register with the DEA.
Although the CSA’s restrictions
governing substances in CIV are not as stringent as those for substances in CI, CII or CIII, they could still limit our ability to market
and commercialize Quilience and Nolazol, if approved for marketing. In addition, failure to maintain compliance with applicable requirements
under the CSA, particularly as manifested in loss or diversion of regulated substances, can result in enforcement action that could include
civil penalties, refusal to renew registrations or quotas, revocation of registrations or quotas or criminal proceedings, any of which
could have a material adverse effect on our business, results of operations and financial condition. Individual states also regulate controlled
substances, and we along with our contract manufacturers will be subject to state regulation on distribution of these products.
Our market is subject to intense competition,
which may result in others commercializing products before or more successfully than us. If we are unable to compete effectively, Quilience
and/or Nolazol may be rendered non-competitive or obsolete, which may adversely affect our operating results.
The development and commercialization
of new products is highly competitive. Our potential competitors include major pharmaceutical companies, specialty pharmaceutical companies
and biotechnology companies worldwide with respect to Quilience and/or Nolazol or any future product candidate that we may seek to develop
or commercialize. Our competitors may succeed in developing, acquiring or licensing technologies and products that are more effective,
have fewer or more tolerable side effects or are more convenient or less costly than Quilience and/or Nolazol or any future product candidate
we may develop, which could render any product candidates obsolete and non-competitive. Our competitors also may obtain FDA or other marketing
approvals for their products before we are able to obtain approval for ours, which could result in competitors establishing a strong market
position before we are able to enter the applicable market.
Many of our potential competitors,
alone or with their strategic partners, have significantly greater financial resources and expertise in research and development, manufacturing,
pre-clinical testing, conducting clinical trials, obtaining marketing approvals and commercializing approved products than we do. There
is a trend toward consolidation in the pharmaceutical and biotechnology industry, and additional mergers and acquisitions in these industries
may result in even more resources being concentrated among a smaller number of our competitors, which may adversely affect us.
Smaller or early-stage companies
may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These
companies also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial
sites and patient registration for clinical trials.
In addition, if we enter the
markets of our product candidates, with such entrance remaining subject to various additional regulatory approvals, too late in the cycle,
we may not achieve commercial success, or we may have to reduce our price in order to effectively compete, which would impact our ability
to generate revenues, obtain profitability and adversely affect our operating results.
While orphan drug product candidates are
typically sold at a high price relative to other medications, the market may not be receptive to high pricing of our product candidates.
We are developing certain
product candidates to treat rare central nervous system, or CNS, disorders with high unmet medical needs, a space where medications are
usually sold at high prices compared with other medications. Accordingly, even if regulatory authorities approve our product candidates,
the market may not be receptive to, and it may be difficult for us to achieve, a per-patient per-year price high enough to allow us to
realize a return on our investment.
Unfavorable global economic conditions,
including as a result of the ongoing war between Russia and Ukraine as well as Israel’s multi-front war, could have a negative impact
on our operations, which could materially and adversely affect our business, financial condition, results of operations, prospects and
market price of our common shares.
Global economic instability
and unfavorable conditions could materially and adversely affect our business. The war between Russia and Ukraine is ongoing. The impact
to Ukraine as well as actions taken by other countries, including new and stricter sanctions imposed by Canada, the United Kingdom, the
European Union, the United States and other countries against officials, individuals, regions, and industries in Russia and Ukraine, and
actions taken by Russia in response to such sanctions, and responses of countries and political bodies to such sanctions, tensions, and
military actions and the potential for more widespread conflict, have resulted in supply chain disruptions, increases in inflation, financial
market volatility and capital markets disruption. In addition, the war between Israel and Hamas is ongoing and has opened additional fronts.
Any resulting instability and unfavorable economic conditions from the wars could disrupt our and our collaborators’ supply chains
and adversely affect our and our collaborators’ ability to conduct ongoing and future clinical trials of our product candidates.
The extent and duration of the wars, sanctions and resulting economic, market and other disruptions are impossible to predict, but could
be substantial. Any such disruptions may heighten the impact of the other risks described in this annual report.
If product liability lawsuits are brought
against us, we may incur substantial liabilities, even if we have appropriate insurance policies, and we may be required to limit commercialization
of our product candidates.
We are exposed to potential
product liability and professional indemnity risks that are inherent in the research, development, manufacturing, marketing and use of
pharmaceutical products. Currently, we have no products that have been approved for marketing or commercialization; however, the use of
our product candidates in clinical trials, and the sale of these product candidates, if approved, in the future, may expose us to liability
claims. Product liability claims may be brought against us or our partners by participants enrolled in our clinical trials, patients,
health care providers, pharmaceutical companies, our collaborators or others using, administering or selling any of our future approved
products. If we cannot successfully defend ourselves against any such claims, we may incur substantial liabilities, even if we have product
liability or such other applicable insurance policies in effect. We may not be able to maintain adequate levels of insurance for these
liabilities at reasonable cost and/or reasonable terms. Excessive insurance costs or uninsured claims would add to our future operating
expenses and adversely affect our financial condition. As a result of such lawsuits and their potential results, we may be required to
limit commercialization of our product candidates. Regardless of the merits or eventual outcome, liability claims may result in:
| |
● |
decreased demand for our product candidates; |
| |
|
|
| |
● |
termination of clinical trial sites or entire trial programs; |
| |
|
|
| |
● |
injury to our reputation and negative media attention; |
| |
● |
product recalls or increased warnings on product labels; |
| |
● |
withdrawal of clinical trial participants; |
| |
● |
costs of to defend the related litigation; |
| |
● |
diversion of management and our resources; |
| |
|
|
| |
● |
substantial monetary awards to, or costly settlements with, clinical trial participants, patients or other claimants; |
| |
|
|
| |
● |
higher insurance premiums; |
| |
|
|
| |
● |
loss of initiation of investigations by regulators or other authorities; and |
| |
|
|
| |
● |
the inability to successfully commercialize our product candidates, if approved. |
Risks Related to the Regulatory Environment
Obtaining approval of an NDA or a MAA even
after clinical trials that are believed to be successful is an uncertain process.
We are not permitted to market
Quilience and/or Nolazol in the United States or the EU until we receive regulatory approval of an NDA from the FDA or MAA from the EMA,
or in any foreign countries until we receive the requisite approval from regulatory authorities in such countries. We have not received
regulatory clearance to conduct the additional clinical trials that are necessary to be able to submit an NDA to the FDA for Nolazol.
Similarly, we have not received regulatory clearance in the EU to conduct clinical trials that are necessary to receive approval of a
MAA for Quilience in Europe. As such, we have not submitted an MAA for any of our product candidates.
We may be able make our products
available on a named patient basis and generate pre-approval revenues from compassionate use activities leveraging on an expanded access
policy in certain countries around the world.
Even if we complete our planned
clinical trials and believe the results to be successful, all of which are uncertain, obtaining regulatory approval is an extensive, lengthy,
expensive and uncertain process, and the FDA and EMA, and other regulatory authorities may delay, limit or deny approval of Quilience
and/or Nolazol for many reasons, including, but not limited to:
| |
● |
we may not be able to demonstrate to their satisfaction that the product candidate is a safe or effective treatment for a given indication; |
| |
|
|
| |
● |
the results of clinical trials may not meet the level of statistical significance or clinical significance required by the regulatory agencies; |
| |
● |
disagreements regarding the number, design, size, conduct or implementation of our clinical trials, or with our interpretation of data from pre-clinical studies or clinical trials; |
| |
|
|
| |
● |
a lack of acceptance of the accuracy or sufficiency of the data generated at our clinical trial sites to demonstrate, among others, that clinical and other benefits outweigh its safety risks or to support the submission of an NDA or MAA; |
| |
|
|
| |
● |
difficulties scheduling an advisory committee meeting in a timely manner or the advisory committee, or such other similar committee, may recommend against approval of our application or may recommend that such regulators require, as a condition of approval, additional pre-clinical studies or clinical trials, limitations on approved labeling, or distribution and use restrictions; |
| |
● |
the requirement that we develop a Risk Evaluation and Mitigation Strategy, or REMS, as a condition of approval, which may or may not be feasible for us; |
| |
● |
the identification of deficiencies in the manufacturing processes or facilities of third-party manufacturers with which we enter into agreements for clinical and commercial supplies; |
| |
|
|
| |
● |
changes in approval policies or the adoption of new regulations by such regulators; and |
| |
|
|
| |
● |
we may be unable to be granted a PIP deferral which we intend to request from the EMA for delayed clinical trials and subsequent approval in children; this may delay our clinical trial program or approvals for adults, or we may have successful clinical trial results for adults but not children (if we were required to conduct pediatric studies prior to the receipt of an NDA or MAA for use of our product candidates in adults), or vice versa. |
Before we can submit an NDA
to the FDA, we must conduct pivotal trials, in addition to human pharmacokinetic and bioavailability studies, that will be substantially
broader than our Phase 2 trial for Nolazol and or Quilience. An NDA must be supported by extensive clinical and pre-clinical data, as
well as extensive information regarding chemistry, manufacturing and controls to demonstrate the safety and effectiveness of the applicable
product candidate. The number and types of pre-clinical studies and clinical trials that will be required varies depending on the product
candidate, the disease or condition that the product candidate is designed to target and the regulations applicable to any particular
product candidate. Obtaining approval of an NDA is a lengthy, expensive and uncertain process, and we may not be successful in obtaining
approval. The FDA review processes can take years to complete and approval is never guaranteed.
In this respect, we will also
need to agree on a protocol with the FDA for the pivotal trials before commencing those trials. Pivotal trials frequently produce unsatisfactory
results even though prior clinical trials were successful. Therefore, the results of the additional trials that we conduct may or may
not be successful. The FDA may suspend all clinical trials or require that we conduct additional clinical, nonclinical, manufacturing
validation or drug product quality studies and submit those data before it will consider or reconsider the NDA. Depending on the extent
of these or any other studies, approval of any applications that we submit may be delayed by several years, or may require us to expend
more resources than we have available. It is also possible that additional studies, if performed and completed, may not be considered
sufficient by the FDA to approve the NDA. If any of these outcomes occur, we would not receive approval for Quilience or Nolazol at such
time, if any, when we seek FDA approval. We may face similar risks with respect to obtaining regulatory approval from the EMA for Quilience,
and for Nolazol, at such time, if any, when we seek EMA approval. The risks that we face in obtaining applicable approvals from the FDA
and EMA for Quilience and/or Nolazol or any other product candidate that we may seek to develop, may also exist with other regulatory
authorities, such as those in Latin America.
Even if we obtain FDA, EMA
or other regulatory approval for Quilience and/or Nolazol, the approval might contain significant limitations related to use restrictions
for certain age groups, warnings, precautions or contraindications, or may be subject to significant post-marketing studies or risk mitigation
requirements. In addition, even if we obtain an MAA from the EMA for the use of Quilience in adults, there can be no guarantee that we
will receive an MAA for Quilience for the use in children. If we are unable to successfully commercialize Quilience and/or Nolazol we
may be forced to cease operations.
The results of clinical trials conducted
at clinical sites outside the United States may not be accepted by the FDA and the results of clinical trials conducted at clinical sites
in the United States may not be accepted by international regulatory authorities.
We are conducting our
Phase 2 clinical trials in the United States. In future, we are planning to conduct Phase 2b and or Phase 3 clinical trials in the
United States and the EU. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of
this data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be well-designed and conducted
and performed by qualified investigators in accordance with ethical principles such as IRB or ethics committee approval and informed
consent. The study population must also adequately represent the U.S. population, and the data must be applicable to the U.S.
population and U.S. medical practice in ways that the FDA deems clinically meaningful. Generally, the subject population for any
clinical trials conducted outside of the United States must be representative of the U.S. population. In addition, while these
clinical trials are subject to the applicable local laws, FDA acceptance of the data will be dependent upon its determination that
the trials were conducted consistent with all applicable U.S. laws and regulations. There can be no assurance the FDA or
international regulatory authorities will accept data from trials conducted outside of the United States or inside the United
States, as the case may be, as adequate support of a marketing application. If the FDA or international regulatory authorities do
not accept the data from sites in our globally conducted clinical trials, it would likely result in the need for additional trials,
which would be costly and time-consuming and could delay or permanently halt the development of one or more of our product
candidates.
Our product candidates may cause undesirable
side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial potential or result
in significant negative consequences following regulatory approval, if obtained.
During the conduct of clinical
trials, patients may experience changes in their health, including illnesses, injuries, discomforts or a fatal outcome. It is possible
that as we develop Quilience and Nolazol, or other product candidates that we may seek to develop, in larger, longer and more extensive
clinical trials as use of our product candidates becomes more widespread if they receive regulatory approval, illnesses, injuries, discomforts
and other adverse events that were observed in earlier clinical trials, as well as conditions that did not occur or went undetected in
previous clinical trials, will be reported by subjects. Many times, side effects are only detectable after investigational products are
tested in larger scale, Phase 2b/Phase 3 clinical trials or, in some cases, after they are made available to patients on a commercial
scale after approval. If additional clinical experience indicates that Quilience and/or Nolazol, or other product candidates that we may
seek to develop, have side effects or cause serious or life-threatening side effects, the development of the product candidate may fail
or be delayed, or, if the product candidate has received regulatory approval, such approval may be revoked or limited.
Additionally, if any of our
product candidates receives marketing approval, the FDA or EMA could require us to adopt a REMS to ensure that the benefits outweigh its
risks, which may include, among other things, a medication guide outlining the risks of the product for distribution to patients, a communication
plan to health care practitioners, and restrictions on how or where the product can be distributed, dispensed or used. Furthermore, if
we or others later identify undesirable side effects caused by Quilience and/or Nolazol, several potentially significant negative consequences
could result, including:
| |
● |
regulatory authorities may suspend or withdraw approvals of such a product candidate; |
| |
● |
regulatory authorities may require additional warnings on the label; |
| |
● |
regulatory authorities may issue negative publicity regarding the affected product, including safety communications; |
| |
● |
we may be required to change the way the product is distributed, dispensed or administered, or conduct additional pre-clinical studies or clinical trials; |
| |
|
|
| |
● |
we may need to voluntarily recall our products; and |
| |
● |
we could be sued and held liable for harm caused to patients. |
Any of these events could
prevent us from achieving or maintaining market acceptance of the affected product candidate and could significantly harm our business,
prospects, financial condition and results of operations.
We will need to obtain FDA approval of any
proposed names for our product candidates that gain marketing approval, and any failure or delay associated with such naming approval
may adversely impact our business.
Any name we intend to
use for our product candidates will require approval from the FDA regardless of whether we have secured a formal trademark
registration from the U.S. Patent and Trademark Office, or the U.S. PTO. The FDA typically conducts a review of proposed product
names, including an evaluation of whether proposed names may be confused with the names of other drug products. The FDA may object
to any product name we submit if it believes the name inappropriately implies medical claims. If the FDA objects to any of our
proposed product names, we may be required to adopt an alternative name for our product candidates, which could result in further
evaluation of proposed names with the potential for additional delays and costs.
Obtaining regulatory approval for clinical
trials of Nolazol in children will be more difficult than obtaining such approvals for adult clinical trials since the requirements for
regulatory approval to conduct pediatric clinical trials are more stringent.
Pediatric drug development
requires additional nonclinical work (such as animal studies in juvenile animals and additional reproductive toxicity work), as well as
staged clinical work in determining safe dosing and monitoring. These additional tasks involve investment of significant additional resources
beyond those needed for approval of the drug for adults. Approval of Nolazol for use in children may be significantly delayed due to these
additional requirements and this may have an adverse effect on the commercial prospects for Nolazol and our ability to generate product
revenues would be delayed, possibly materially. We cannot guarantee that we will receive any regulatory approvals to commercialize our
product candidates in children.
Changes in regulatory requirements and guidance
or unanticipated events during our clinical trials may occur, which may result in necessary changes to clinical trial protocols, which
could result in increased costs to us, delay our development timeline or reduce the likelihood of successful completion of our clinical
trials.
Changes in regulatory requirements
and guidance or unanticipated events during our clinical trials may occur, as a result of which we may need to amend clinical trial protocols.
Amendments may require us to resubmit our clinical trial protocols to IRBs for review and approval, which may impact the cost, timing
or successful completion of a clinical trial. If we experience delays in completion of, or if we terminate, any of our clinical trials,
the commercial prospects for Quilience and Nolazol would be harmed and our ability to generate product revenues would be delayed, possibly
materially.
Our development and regulatory strategy
for our product candidates depends in part on published scientific literature and the FDA’s prior findings regarding the safety
and efficacy of approved products containing mazindol. If the FDA does not conclude that our product candidates satisfy the requirements
for the Section 505(b)(2) regulatory approval pathway, or if the requirements for our product candidates under Section 505(b)(2) are not
as we expect, the approval pathway would likely take significantly longer, cost significantly more and entail significantly greater complications
and risks than anticipated and in either case may not be successful.
The Drug Price Competition
and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Amendments, added Section 505(b)(2) to the Federal Food, Drug,
and Cosmetic Act, or FDCA, or Section 505(b)(2). Section 505(b)(2) permits the submission of an NDA where at least some of the information
required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of
reference. We intend to seek FDA approval through the Section 505(b)(2) regulatory pathway for Quilience and may seek this regulatory
pathway for other product candidates that we seek to develop.
If the FDA does not
allow us to pursue the Section 505(b)(2) regulatory pathway as anticipated, we may need to conduct additional pre-clinical and or
clinical trials, provide additional data and information and meet additional standards for regulatory approval. If this were to
occur, the time and financial resources required to obtain FDA approval, and complications and risks associated with FDA approval,
would substantially increase. We may need to obtain additional funding, which could result in significant dilution to the ownership
interests of our then existing shareholders to the extent we issue equity securities or convertible debt. We cannot assure you that
we would be able to obtain such additional financing on terms acceptable to us, if at all. Moreover, inability to pursue the Section
505(b)(2) regulatory pathway could result in new competitive product candidates reaching the market faster than our product
candidates, which could materially adversely impact our competitive position and prospects. Even if we are allowed to pursue the
Section 505(b)(2) regulatory pathway, we cannot assure you that our product candidates will receive the requisite approvals for
commercialization.
We may seek designations for our product
candidates with the FDA and other comparable regulatory authorities that are intended to confer benefits such as a faster development
process or an accelerated regulatory pathway, but there can be no assurance that we will successfully obtain such designations. In addition,
even if one or more of our product candidates are granted such designations, we may not be able to realize the intended benefits of such
designations.
The FDA, and other comparable
regulatory authorities, offer certain designations for product candidates that are intended to encourage the research and development
of pharmaceutical products addressing conditions with significant unmet medical need. These designations may confer benefits such as additional
interaction with regulatory authorities, a potentially accelerated regulatory pathway and priority review. There can be no assurance that
we will successfully obtain such designation for Quilience and/or Nolazol. In addition, while such designations could expedite the development
or approval process, they generally do not change the standards for approval. Even if we obtain such designations for one or more of our
product candidates, there can be no assurance that we will realize their intended benefits.
For example, we may seek a
Breakthrough Therapy designation from the FDA for one or more of our product candidates. A Breakthrough Therapy designation is defined
as a therapy that is intended, alone or in combination with one or more other therapies, to treat a serious or life-threatening disease
or condition, if preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies
on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For therapies
that have Breakthrough Therapy designation, interaction and communication between the FDA and the sponsor of the trial can help to identify
the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Therapies
with Breakthrough Therapy designation from the FDA are also eligible for accelerated approval. Designation as a Breakthrough Therapy is
within the discretion of the FDA. Accordingly, even if we believe one of our product candidates meets the criteria for Breakthrough Therapy
designation, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a Breakthrough Therapy
designation for a product candidate may not result in a faster development process, review or approval compared to therapies considered
for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our
product candidates qualify for Breakthrough Therapy designation, the FDA may later decide that such product candidates no longer meet
the conditions for qualification.
We may also seek Fast Track
designation from the FDA for some of our product candidates. If a therapy is intended for the treatment of a serious or life-threatening
condition and the therapy demonstrates the potential to address unmet medical needs for this condition, the therapy sponsor may apply
for Fast Track designation. The FDA has broad discretion whether or not to grant this designation, so even if we believe a particular
product candidate is eligible for this designation, there can be no assurance that the FDA would decide to grant it. Even if we do receive
Fast Track designation, we may not experience a faster development process, review or approval compared to conventional FDA procedures,
and receiving a Fast-Track designation does not provide assurance of ultimate FDA approval. The FDA may withdraw Fast Track designation
if it believes that the designation is no longer supported by data from our clinical development program.
We may also seek a priority
medicines scheme, or PRIME, designation for some of our product candidates. PRIME was launched by EMA in 2016 to facilitate the development
of medicines that target an unmet medical need. Through PRIME, the EMA offers early and proactive support to medicine developers with
the aim of optimizing the generation of robust data on a medicine’s benefits and risks and enabling accelerated assessment of medicines
applications. The overall goal is to ensure patients benefit as early as possible from therapies that may significantly improve their
quality of life. PRIME focuses on improving the design of clinical trials in order to ensure the efficient generation of the necessary
clinical data for inclusion in a Marketing Authorization Application, or MAA.
Even if we obtain regulatory approval for
a product candidate, our products will remain subject to ongoing regulatory oversight.
Even if we obtain any regulatory
approval for our product candidates, they will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage,
advertising, promotion, sampling, record-keeping and submission of safety and other post-market information. Any regulatory approvals
that we receive for our product candidates also may be subject to a REMS, limitations on the approved indicated uses for which the product
may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase
4 clinical trials, and surveillance to monitor the quality, safety and efficacy of the product. The holder of an approved marketing application
also must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling
or manufacturing process. Advertising and promotional materials must comply with FDA rules and are subject to FDA review, in addition
to other potentially applicable federal and state laws.
In addition, product manufacturers
and their facilities are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory
authorities for compliance with current good manufacturing practices, or cGMP, requirements and adherence to commitments made in the NDA
or foreign marketing application. If a regulatory authority discovers previously unknown problems with a product, such as adverse events
of unanticipated severity or frequency, or problems with the facility where the product is manufactured or disagrees with the promotion,
marketing or labeling of that product, a regulatory authority may impose restrictions relative to that product, the manufacturing facility
or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. If we fail to comply with
applicable regulatory requirements, a regulatory authority may:
| |
● |
issue an untitled letter or warning letter that we are in violation of the law; |
| |
● |
seek an injunction or impose administrative, civil or criminal penalties or monetary fines; |
| |
● |
suspend or withdraw regulatory approval; |
| |
● |
suspend any ongoing clinical trials; |
| |
● |
refuse to approve pending applications or supplements to applications; |
| |
● |
restrict the marketing or manufacturing of the product; |
| |
● |
seize or detain the products or require the withdrawal of the product from the market; |
| |
● |
refuse to permit the import or export of the products; or |
| |
● |
refuse to allow us to enter into supply contracts, including government contracts. |
Any government investigation
of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity.
The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and adversely affect
our business, financial condition, results of operations and prospects.
The FDA and other regulatory authorities
actively enforce the laws and regulations prohibiting the promotion of off-label uses.
If any of our product
candidates are approved and we are found to have improperly promoted off-label uses of those products, we may become subject to
significant liability. The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about
prescription products, if approved. In particular, while the FDA permits the dissemination of truthful and non-misleading
information about an approved product, a manufacturer may not promote a product for uses that are not approved by the FDA or such
other regulatory agencies as reflected in the product’s approved labeling. If we are found to have promoted such off-label
uses, we may become subject to significant liability. The federal government has levied large civil and criminal fines against
companies for alleged improper promotion of off-label use and has enjoined several companies from engaging in off-label promotion.
The FDA has also requested that companies enter into consent decrees, corporate integrity agreements or permanent injunctions under
which specified promotional conduct must be changed or curtailed. If we cannot successfully manage the promotion of our product
candidates, if approved, we could become subject to significant liability, which would materially adversely affect our business and
financial condition.
Obtaining and maintaining regulatory approval
of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product
candidates in other jurisdictions. Our failure to obtain regulatory approval in foreign jurisdictions would prevent our product candidates
from being marketed abroad, and any approval we are granted for our product candidates in the United States would not assure approval
of product candidates in foreign jurisdictions.
In order to market any products
outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding
clinical trial design, safety and efficacy. The research, testing, manufacturing, labeling, approval, sale, marketing and distribution
of drugs are subject to extensive regulation by the FDA in the United States and other regulatory authorities in other countries. These
regulations differ from country to country. Even if we obtain and maintain regulatory approval of our product candidates in one jurisdiction,
such approval does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, but a failure
or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others.
For example, even if the FDA grants marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions
must also approve the manufacturing, marketing and promotion of the product candidate in those countries.
Approval procedures vary among
jurisdictions and can involve requirements and administrative review periods different from those in the United States, including additional
pre-clinical studies or clinical trials as investigations conducted in one jurisdiction may not be accepted by regulatory authorities
in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before
it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to
approval. These regulatory procedures can result in substantial delays in such countries. In other countries, product approval depends
on showing superiority to an approved alternative therapy. This can result in significant expenses for conducting complex clinical trials.
Finally, we do not have any
products approved for sale in any jurisdiction, including international markets, and we do not have experience in obtaining regulatory
approval. If we, or any third parties with whom we work, fail to comply with regulatory requirements in the United States or international
markets or to obtain and maintain required approvals or if regulatory approvals in international markets are delayed, our target market
may be reduced and our ability to realize the full market potential of our products will likely be harmed. The inability to meet continuously
evolving regulatory standards for approval may result in our failing to obtain regulatory approval to market our current product candidates,
which could significantly harm our business, results of operations and prospects.
We may be unable to obtain or maintain orphan
drug designation for our product candidates and, even if we obtain such designation, we may not be able to realize the benefits of such
designation, including potential marketing exclusivity of our product candidates, if approved.
Regulatory authorities
in some jurisdictions, including the United States and EU, may designate drugs for relatively small patient populations as
“orphan drugs.” Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug intended to
treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals in the
United States or a patient population of greater than 200,000 individuals in the United States, but for which there is no reasonable
expectation that the cost of developing the drug will be recovered from sales in the United States. In the EU, the European
Commission grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or
treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the EU
community. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a
life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of
the drug in the EU would be sufficient to justify the necessary investment in developing the drug.
The FDA granted orphan drug
designation to mazindol for the treatment of narcolepsy in the United States. We also received orphan drug designation for mazindol in
the EU for the treatment of IH. This designation of a product candidate as an orphan product does not mean that any regulatory agency
will accelerate regulatory review of, or ultimately approve, that product candidate, nor does it limit the ability of any regulatory agency
to grant orphan drug designation to product candidates of other companies that treat the same indications as our product candidates prior
to our product candidates receiving exclusive marketing approval.
Generally, if a product candidate
with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation,
the drug may be entitled to a period of marketing exclusivity, which precludes the FDA or the EMA from approving another marketing application
for the same drug for that time period, except in limited circumstances. If another sponsor receives such approval before we do (regardless
of our orphan drug designation), we will be precluded from receiving marketing approval for our product for the applicable exclusivity
period. The applicable period is seven years in the United States and ten years in the EU, which may be extended by six months and two
years, respectively, in the case of product candidates that have complied with the respective regulatory agency’s agreed upon PIP.
The exclusivity period in the EU can be reduced to six years if a drug no longer meets the criteria for orphan drug designation or if
the drug is sufficiently profitable so that market exclusivity is no longer justified. Orphan drug exclusivity may be revoked if any regulatory
agency determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity
of the drug to meet the needs of patients with the rare disease or condition.
Even if we obtain orphan drug
exclusivity for a product candidate, that exclusivity may not effectively protect the product candidate from competition because different
drugs can be approved for the same condition. Even after an orphan drug is approved, the FDA may subsequently approve another drug for
the same condition if the FDA concludes that the latter drug is not the same drug or is clinically superior in that it is shown to be
safer, more effective or makes a major contribution to patient care. In the EU, marketing authorization may be granted to a similar medicinal
product for the same orphan indication if:
| |
● |
the second applicant can establish in its application that its medicinal product, although similar to the orphan medicinal product already authorized, is safer, more effective or otherwise clinically superior; |
| |
● |
the holder of the marketing authorization for the original orphan medicinal product consents to a second orphan medicinal product application; or |
| |
● |
the holder of the marketing authorization for the original orphan medicinal product cannot supply sufficient quantities of orphan medicinal product. |
Risks Related to Our Financial Condition and
Capital Requirements
Our financial statements for the year ended
December 31, 2024, contained a disclosure in Note 1 regarding substantial doubt about our ability to continue as a going concern. This going
concern disclosure in Note 1 of financial statements could prevent us from obtaining new financing on reasonable terms or at all and risk
our ability to continue operating as a going concern.
To date, the Company has not
generated significant revenues from its activities and has incurred substantial operating losses. In part because we have incurred
losses in each year since our inception, our audited financial statements for the period ended December 31, 2024 contain a disclosure
in Note 1 regarding substantial doubt about our ability to continue as a going concern. These events and conditions, along with other
matters, indicated that a material uncertainty existed as of December 31, 2024, that raises substantial doubt on our ability to continue
as a going concern. The financial statements for the year ended December 31, 2024 have been prepared assuming that we will continue
as a going concern. Management expects the Company to continue to generate substantial operating losses and to continue to fund
its operations primarily through the utilization of its current financial resources, sales of its products, and through additional raises
of capital. As of December 31, 2024, we incurred accumulated losses of approximately $74.4 million. The disclosure regarding substantial
doubt about our ability to continue as a going concern could materially limit our ability to raise additional funds through the issuance
of equity or debt securities or otherwise in the future. Further financial statements may also include a similar disclosure with respect
to our ability to continue as a going concern. Until we can generate significant recurring revenues, we expect to satisfy our future
cash needs through existing cash, debt or equity financing. We expect to require additional financing to fund our operations in the near
future. We cannot be certain that additional funding will be available to us on acceptable terms, if at all. If funds are not available,
we may be required to delay, reduce the scope of, or eliminate research or development plans for, or commercialization efforts with respect
to our products and it risks our ability to continue operating as a going concern.
We are exposed to various risks related
to legal or administrative proceedings or claims that could adversely affect our financial condition, results of operations and reputation,
and may cause loss of business.
Litigation in general can
be expensive, lengthy and disruptive to normal business operations. Moreover, the results of complex legal proceedings are difficult to
predict. We and/or our directors and officers may be involved in allegations, litigation or legal or administrative proceedings from time
to time.
On August 27, 2024, the Company
received correspondence from Université de Lausanne, initiating the official “audience de conciliation” procedure,
overseen by the ordinary civil court in Lausanne. The hearing was scheduled for October 9, 2024, at the Tribunal d’arrondissement
de Lausanne. The complaint pertains to an unpaid invoice for research services amounting to $110,179, plus interest at a rate of 5%. At
the hearing on October 9, 2024, Université de Lausanne was not open to discussing a potential settlement. The Company asserts that
the services provided did not meet the required standard of care and intends to defend its position. On 9 May 2025, the Company filed
a statement of defense and asserted a counterclaim in CHF 30,000 plus compensatory interests at a rate of 5% per annum since June 29,
2022.
Regardless of the merits,
responding to allegations, litigation or legal or administration proceedings and defending against litigation can be time-consuming and
costly, and may result in us incurring substantial legal and administrative expenses, as well as divert the attention of our management.
Any such allegations, lawsuits or proceedings could have a material adverse effect on our business operations. Further, unfavorable outcomes
from these claims or lawsuits could adversely affect our business, financial condition and results of operations.
We believe that our current cash on hand
and access to existing financing arrangements will not be sufficient to fund our projected operating requirements for a period of one
year from the issuance of the financial statements included elsewhere in this annual report. This raises substantial doubt about our ability
to continue as a going concern. There can be no assurance that these funds will be available, or if they are available, their availability
will be on terms acceptable to the Company or in an amount sufficient to enable the Company to fully complete its development activities,
sustain operations or pay back its debt.
We believe that our
current cash on hand and access to existing financing arrangements will not be sufficient to fund our projected operating
requirements for a period of one year from the issuance of the financial statements included elsewhere in this annual report. This
raises substantial doubt about our ability to continue as a going concern. We expect that we will be required to obtain additional
liquidity in order to fund our operations through the approval of our lead product candidate. Until we can generate significant
revenues, if ever, we expect to satisfy our future cash needs through debt, equity financing or income from a partnership deal. We
cannot be certain that funding will be available to us on acceptable terms, if at all. If funds are not available, we may be
required to delay, reduce the scope of, or eliminate research or development plans for, or commercialization efforts with respect to
our products. We may ultimately be forced to file for bankruptcy if no funds are available either through a financing or a strategic
partnership.
Until we can generate significant
revenues, if ever, we expect to satisfy our future cash needs through debt or equity financing. We cannot be certain that additional funding
will be available to us on acceptable terms, if at all. We believe that our current cash on hand and access to existing financial arrangements
will not be sufficient to fund our projected operating requirements for at least one year from the issuance of the financial statements
included elsewhere in this annual report. If funds are not available, we may be required to delay, reduce the scope of, or eliminate research
or development plans for, or commercialization efforts with respect to our products.
We have a limited operating history and
we have incurred significant operating losses since our inception, and anticipate that we will incur continued losses for the foreseeable
future.
We are an emerging biopharmaceutical company with a limited operating
history. To date, we have focused almost exclusively on developing product candidates using the active ingredient mazindol in proprietary
formulations. We have funded our operations to date primarily through proceeds from the private placement of common shares, convertible
instruments, related party credit facilities, shareholder loans, an initial public offering of common shares and Warrants, and drawdowns
from a standby equity distribution agreement. We have only a limited operating history upon which you can evaluate our business and prospects.
In addition, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties
frequently encountered by companies in new and rapidly evolving fields, particularly in the pharmaceutical industry. To date, although
we received an upfront payment of approximately $2.5 million pursuant to the EF License Agreement (defined below) in 2019, we have not
generated revenue from the sale of our product candidates (see “Item 5. Operating and Financial Review and Prospects — Components
of Operating Results — Licensing Agreement” for additional information). We have incurred losses in each year since our
inception. Our net loss attributable to holders of our common shares for the year ended December 31, 2024 was approximately $4.1 million
and for the year ended December 31, 2023 was approximately $12.2 million. As of December 31, 2024, we had an accumulated deficit of approximately
$74.4 million. Substantially all of our operating losses resulted from costs incurred in connection with our clinical development program
and from general and administrative costs associated with our operations.
We expect our research and
development expenses to increase in connection with our planned expanded clinical trials. In addition, if we obtain marketing approval
for Quilience and/or Nolazol, or any other current or future product candidate, we will likely incur significant sales, marketing and
outsourced manufacturing expenses, as well as continued research and development expenses. Furthermore, we expect to incur additional
costs associated with operating as a public company, which we estimate will be at least several hundred thousand dollars annually. As
a result, we expect to continue to incur significant and increasing operating losses for the foreseeable future. Because of the numerous
risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or
when we will become profitable, if at all.
We expect to continue to incur
significant losses until we are able to commercialize our product candidates, which we may not be successful in achieving. We anticipate
that our expenses will increase substantially if and as we:
| |
● |
continue the research and development of our product candidates; |
| |
● |
expand the scope of our current clinical studies for our product candidates; |
| |
● |
seek regulatory and marketing approvals for our product candidates that successfully complete clinical studies; |
| |
● |
establish a sales, marketing, and distribution infrastructure to commercialize our product candidates; |
| |
● |
seek to identify, assess, acquire, license, and/or develop other product candidates and subsequent generations of our current product candidates; |
| |
● |
seek to maintain, protect, and expand our intellectual property portfolio; |
| |
● |
seek to attract and retain skilled personnel; and |
| |
● |
create additional infrastructure to support our operations as a public company and our product candidate development and planned future commercialization efforts. |
We have not generated revenues from any
product candidate and may never be profitable.
Our ability to become profitable
depends upon our ability to generate revenues. To date, we have not generated any revenue from our pre-clinical and clinical development
stage product candidates, Quilience, Nolazol and NLS-4, and we do not know when, or if, we will generate any such revenue. We do not expect
to generate significant revenues unless or until we obtain marketing approval of, and commercialize, Quilience and/or Nolazol and/or the
Aexon platform that we may seek to develop in the future. Our ability to generate future revenues from product candidate sales depends
heavily on our success in many areas, including but not limited to:
| |
● |
obtaining favorable results from and progress the pre-clinical and clinical development of our product candidates, namely Quilience and/or Nolazol; |
| |
● |
developing and obtaining regulatory approval for registration studies protocols for our product candidates, namely Quilience and/or Nolazol; |
| |
● |
subject to successful completion of registration and clinical trials of Quilience and/or Nolazol, applying for and obtaining marketing approval; |
| |
● |
establishing and maintaining supply and manufacturing relationships with third parties that can provide adequate (in amount and quality) products, and at acceptable costs, to support market demand for our product candidates, if marketing approval is received; |
| |
● |
identifying, assessing, acquiring and/or developing new product candidates; |
| |
● |
accurately identifying demand for our product candidates; |
| |
● |
continued consumer interest in treatments to the symptoms of narcolepsy, IH, ADHD and Long-COVID; |
| |
● |
obtaining market acceptance of our product candidates, if approved for marketing, as viable treatment options; |
| |
● |
negotiating favorable terms in any collaboration, licensing or other arrangements into which we may enter; |
| |
● |
obtaining and maintaining CIV labeling (no FDA imposed boxed warning, commonly referred to as a “Black Box” warning) of our lead product candidate Quilience and follow-on product candidates, Nolazol and NLS-4; |
| |
● |
establishing and nurturing relationships with the leading prescribers of narcolepsy, ADHD and Long-COVID prescriptions in the United States; and |
| |
● |
attracting, hiring, and retaining qualified personnel. |
Alexander Zwyer, our Chief Executive Officer,
and Eric Konofal, our Chief Scientific Officer, have interests that may be different from, or in addition to, the interests of our shareholders
and the Company and Aexon may not have sufficient funds to continue or grow operations.
Alexander Zwyer owns 35% of
Aexon, and Eric Konofal owns 59% of Aexon. Mr. Konofal is the founder of Aexon, with which we have a license agreement, and also serves
as the President of Aexon. Mr. Zwyer holds no board or executive position at Aexon Labs, Inc., or Aexon. Mr. Zwyer and Mr. Konofal may
have interests in the transactions with Aexon that may be different from, or in addition to, the interests of our shareholders and that
may create potential conflicts of interest. Each of us and Aexon expect to be required to obtain additional liquidity in order to fund
operations through the approval of certain of each of the company’s products. Until each company can generate significant revenues,
if ever, each company expects to satisfy its future cash needs through debt or equity financings. Each company cannot be certain that
funding will be available to it on acceptable terms, if at all. If funds are not available, each company may be required to delay, reduce
the scope of, or eliminate research or development plans for, or commercialization efforts with respect to each company’s products.
We expect that we will need to raise substantial
additional funding before we can expect to complete the development of Quilience or any other product candidate. This additional financing
may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit
or terminate our product candidate development efforts or other operations.
As of December 31, 2024, our
cash and cash equivalents were approximately $1.7 million, and we had a positive working capital of $1.4 million and an accumulated deficit
of $74.4 million. Based upon our currently expected level of operating expenditures, we believe that our current cash on hand and access
to existing financial arrangements will not be sufficient to fund our projected operating requirements for a period of one year from the
issuance of these financial statements included elsewhere in this annual report. This raises substantial doubt about our ability to continue
as a going concern. We expect that we will require substantial additional capital to commercialize our product candidates and put in place
multiple options to raise the funds necessary to support our operations. However, our operating plans may change as a result of many factors
that may currently be unknown to us, and we may need to seek additional funds sooner than planned. Our future funding requirements will
depend on many factors, including but not limited to:
| |
● |
our clinical trial results and the costs for conducting pivotal trials; |
| |
● |
the cost, timing and outcomes of seeking marketing approval of Quilience and/or Nolazol; |
| |
● |
the cost of filing and prosecuting patent applications and the cost of defending our patents; |
| |
● |
the cost of prosecuting patent infringement actions against third parties; |
| |
● |
development of other early-stage development product candidates; |
| |
● |
the costs associated with commercializing Quilience and/or Nolazol if we receive marketing approval, including the cost and timing of establishing sales and marketing capabilities to market and sell Quilience and/or Nolazol; |
| |
● |
subject to receipt of marketing approval, revenue received from sales of approved products, if any, in the future; |
| |
● |
any product liability or other lawsuits related to our products; |
| |
● |
the expenses needed to attract and retain skilled personnel; and |
| |
● |
the costs associated with being a public company. |
Any additional fundraising
efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize
our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable
to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of holders of our securities and
the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price
of our common shares or our publicly traded Warrants to decline. The incurrence of indebtedness could result in increased fixed payment
obligations, and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional
debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could
adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborative
partners or otherwise at an earlier stage than otherwise would be desirable, and we may be required to relinquish rights to some of our
technologies or product candidates or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our
business, operating results and prospects. Even if we believe that we have sufficient funds for our current or future operating plans,
we may seek additional capital if market conditions are favorable or if we have specific strategic considerations.
If we are unable to obtain
funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development
programs or the development or commercialization, if any, of any product candidates or be unable to expand our operations or otherwise
capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
Risks Related to Our Reliance on Third Parties
We are dependent on a sole manufacturer
for mazindol. Any delay, price increase or unavailability of mazindol could materially adversely affect our ability to conduct clinical
trials and, if this were to occur after we obtained commercialization and marketing approval, could cause us to cease operations.
Currently we rely on a single
manufacturer for mazindol for our clinical trials conducted to date pursuant to purchase orders and do not have any existing contract
with such manufacturer for future supplies. The FDA requires identification of raw material suppliers in applications for approval of
drug products. If mazindol were to be unavailable from the specified manufacturer, FDA approval of a new manufacturer, assuming one is
found, could delay the manufacture of the drug involved or delay any clinical trial we are then conducting or planning to conduct. Either
such occurrence could have an adverse effect on our operations and reputation or could cause us to cease operations.
Furthermore, there is a risk
of a sole approved manufacturer significantly raising prices. If prices for mazindol or other raw materials were to be significantly increased,
our profit margins and sales, if any, would be greatly reduced and, assuming our products were approved for commercialization or marketing,
delay product launches, or delay clinical trials at earlier stages of development. Such price increase occurrences could be resolved by
the successful FDA approval of an alternate supplier; however, such approval process can be lengthy and costly. There can be no guarantee
that a resolution would be reached in the event of a significant price increase by our supplier of mazindol.
We rely on third parties to conduct our
pre-clinical and clinical studies and perform other tasks for us. If these third parties do not successfully carry out their contractual
duties, meet expected deadlines or comply with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize
our product candidates and our business could be substantially harmed.
We have relied upon and
plan to continue to rely upon third-party CROs to monitor and manage data for our ongoing pre-clinical and clinical programs. We
rely on these parties for execution of our pre-clinical and clinical studies, and control only certain aspects of their activities.
Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol,
legal, regulatory and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities. We
and our CROs and other vendors are required to comply with current cGMP, Good Clinical Practices, or GCP, quality system
requirements, or QSR, and Good Laboratory Practices, or GLP, which are regulations and guidelines enforced by the FDA, the Competent
Authorities of the Member States of the European Economic Area, and comparable foreign regulatory authorities for all of our product
candidates in clinical development. Regulatory authorities enforce these regulations through periodic inspections of study sponsors,
principal investigators, study sites and other contractors. If we or any of our CROs or vendors fail to comply with applicable
regulations, the clinical data generated in our clinical studies may be deemed unreliable and the FDA, EMA or comparable foreign
regulatory authorities may require us to perform additional clinical studies before approving our marketing applications. We cannot
assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical
studies comply with GCP regulations. In addition, our clinical studies must be conducted with product candidates which are produced
under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical studies, which would delay
the regulatory approval process.
We have no manufacturing capacity and anticipate
reliance on third-party manufacturers for our products.
We do not currently operate
manufacturing facilities for clinical production of Quilience and/or Nolazol. We do not intend to develop facilities for the manufacture
of products for clinical trials or commercial purposes in the foreseeable future. We will rely on third-party manufacturers to produce
bulk drug products required for our clinical trials and commercial sales, if any. We plan to continue to rely upon contract manufacturers
and, potentially, collaboration partners to manufacture commercial quantities of our drug product candidates if and when approved for
marketing by the applicable regulatory authorities. Our contract manufacturers have not completed process validation for the drug substance
manufacturing process. Process validation involves a series of activities taking place over the lifecycle of the product and process.
If our contract manufacturers are not approved by the FDA, or other regulatory bodies, our commercial supply of drug substance will be
significantly delayed and may result in significant additional costs. If the FDA does not consider the result of the process validation
or required testing to be satisfactory, regulatory approval and/or commercial supply after launch may be delayed. The FDA and similar
foreign regulatory bodies may also implement new requirements, or change their interpretation and enforcement of existing requirements,
for manufacturers, packaging or testing of products at any time. If we, or our contract manufacturers are unable to comply with such process
validation or other requirements, we may be subject to regulatory, civil actions or penalties which could harm our business.
We purchase finished mazindol,
the molecule in both of Quilience and Nolazol, from a single third party, currently under a development agreement with us. In addition,
we do not have an agreement in place for, but we have identified, a secondary fill/finish supplier. There can be no guarantee that we
will enter into an agreement with such fill/finish supplier or that an effort to enter into such an agreement will be on favorable terms.
If we need to identify an additional fill/finish manufacturer, we would not be able to do so without delay and likely significant additional
cost.
Our existing manufacturer
and any future contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business. In the event
of a natural disaster, business failure, strike or other difficulty, such as difficulties involving production yields, quality control
and quality assurance, we may be unable to replace a third-party manufacturer in a timely manner and the production of any product candidate
or commercialized drug would be interrupted, resulting in delays and additional costs.
In addition, because the contract
manufacturer of our drug substance is located in the United States, we may face difficulties in importing our drug substances into Europe
as a result of, among other things, import inspections, incomplete or inaccurate import documentation or defective packaging.
We and our collaborators and contract manufacturers
are subject to significant regulation with respect to manufacturing our product candidates. The manufacturing facilities on which we rely
may not continue to meet regulatory requirements and have limited capacity.
Manufacturers and their facilities
are required to comply with extensive regulatory requirements, including ensuring that quality control and manufacturing procedures conform
to cGMPs. These cGMP regulations cover all aspects of manufacturing relating to our product candidates. These regulations govern manufacturing
processes and procedures (including record keeping) and the implementation and operation of quality systems to control and assure the
quality of investigational product candidates and products approved for sale. Poor control of production processes can lead to the introduction
of contaminants or to inadvertent changes in the properties or stability of our product candidates that may not be detectable in final
product testing. We, our collaborators or our contract manufacturers must supply all necessary documentation in support of an NDA or MAA
on a timely basis and must adhere to GLP and cGMP QSR regulations enforced by the FDA and other regulatory authorities through their facilities
inspection program. Some of our contract manufacturers have never produced a commercially approved pharmaceutical product and therefore
have not obtained the requisite regulatory authority approvals to do so. The facilities and quality systems of some or all of our collaborators
and third-party contractors must pass a pre-approval inspection for compliance with the applicable regulations as a condition of regulatory
approval of our product candidates or any of our other potential product candidates. In addition, the regulatory authorities may, at any
time, audit or inspect a manufacturing facility involved with the preparation of our product candidates or our other potential product
candidates or the associated quality systems for compliance with the regulations applicable to the activities being conducted. We do not
control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with the regulatory
requirements. If these facilities do not pass a pre-approval plant inspection, regulatory approval of the product candidates may not be
granted or may be substantially delayed until any violations are corrected to the satisfaction of the regulatory authority, if ever. Moreover,
if our contract manufacturers fail to achieve and maintain high manufacturing standards, in accordance with applicable regulatory requirements,
or there are substantial manufacturing errors, this could result in patient injury or death, product shortages, product recalls or withdrawals,
delays or failures in product testing or delivery, cost overruns or other problems that could seriously harm our business.
Any collaboration arrangements that we may
enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our current and
potential future product candidates.
We may seek collaboration
arrangements with pharmaceutical or biotechnology companies for the development or commercialization of our current and potential future
product candidates. We may enter into these arrangements on a selective basis depending on the merits of retaining commercialization rights
for ourselves as compared to entering into selective collaboration arrangements with other pharmaceutical or biotechnology companies for
each product candidate, both in the United States and internationally. We will face, to the extent that we decide to enter into collaboration
agreements, significant competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming
to negotiate, document and implement. We may not be successful in our efforts to establish and implement collaborations or other alternative
arrangements should we so choose to enter into such arrangements. The terms of any collaborations or other arrangements that we may establish
may not be favorable to us.
Disagreements between parties
to a collaboration arrangement regarding clinical development and commercialization matters can lead to delays in the development process
or commercializing the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements
can be difficult to resolve if neither of the parties has final decision-making authority.
Collaborations with pharmaceutical
or biotechnology companies and other third parties often are terminated or allowed to expire by the other party. Any such termination
or expiration could adversely affect us financially and could harm our business reputation.
Our reliance on third parties requires us
to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated
or disclosed.
Because we rely on third parties
to develop and manufacture our product candidates, we must, at times, share trade secrets with them. We seek to protect our proprietary
technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research
agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning
research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our
confidential information, such as trade secrets. Despite the contractual provisions employed when working with third parties, the need
to share trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors,
are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our
proprietary position is based, in part, on our know-how and trade secrets, a competitor’s discovery of our trade secrets or other
unauthorized use or disclosure would impair our competitive position and may have a material adverse effect on our business.
Risks Related to Our Intellectual Property
We have been granted several issued patents
and filed multiple patent applications. There can be no assurance that any of our patent applications will result in issued patents. As
a result, our ability to protect our proprietary technology in the marketplace may be limited.
We have been granted issued
patents in countries worldwide. These issued patents cover a range of areas including: the therapeutic use of mazindol for the treatment
of ADHD and combination therapies containing mazindol (add-ons with iron or stimulants) in sleep disorders such as narcolepsy or IH. We
have also filed patent applications in many countries worldwide. Unless and until our pending patent applications are issued, their protective
scope is impossible to determine. It is impossible to predict whether or how many of our patent applications will result in issued patents.
Even if pending applications are issued, they may be issued with coverage significantly narrower than what we currently seek.
Our proprietary position for our product
candidates currently depends upon patents protecting the method of use, which may not prevent a competitor or other third party from using
the same product candidate for another use.
The primary patent based intellectual
property protection for our product candidates are patents granted on the method of use and formulation. We do not have patents or patent
applications covering our products as a composition of matter (i.e., compound claims) for all our product candidates.
Composition of matter patent
claims on the active pharmaceutical ingredient, or API, in pharmaceutical drug products are generally considered to be the favored form
of intellectual property protection for pharmaceutical products, as such patents provide protection without regard to any particular method
of use, manufacture or formulation of the API used. Method of use patent claims protect the use of a product for the specified method
and dosing. These types of patent claims do not prevent a competitor or other third party from making and marketing an identical API for
an indication that is outside the scope of the method claims or from developing a different dosing regimen. Moreover, even if competitors
or other third parties do not actively promote their product for our targeted indications or uses for which we may obtain patents, physicians
may recommend that patients use these products off-label, or patients may do so themselves. Although off-label use may infringe or contribute
to the infringement of method of use patents, the practice is common and such infringement is difficult to prevent or prosecute.
Because the patent positions of pharmaceutical
products are complex and uncertain, we cannot predict the scope and extent of patent protection of our issued patents for our product
candidates.
Our issued patents may not
ensure the protection of our intellectual property for a number of reasons, including without limitation the following:
| |
● |
any issued patents may not be broad or strong enough to prevent competition from other drug products including identical or similar drug products; |
| |
● |
if issued patents expire, there would be no protections against competitors making generic equivalents; |
| |
● |
there may be prior art of which we are not aware that may affect the validity or enforceability of a patent claim; |
| |
● |
there may be other patents existing, now or in the future, in the patent landscape for Quilience and Nolazol, or any other product candidates that we seek to commercialize or develop, if any, that will affect our freedom to operate; |
| |
● |
if our patents are challenged, a court could determine that they are not valid or enforceable; |
| |
● |
a court could determine that a competitor’s technology or product does not infringe our patents; |
| |
● |
our patents could irretrievably lapse due to failure to pay fees or otherwise comply with regulations, or could be subject to compulsory licensing; and |
| |
● |
if we encounter delays in our development or clinical trials, the period of time during which we could market our products under patent protection would be reduced. |
Obtaining and maintaining our patent protection
depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent
agencies, and our patent protection could be reduced or eliminated for noncompliance with these requirements.
Periodic maintenance fees
on any issued patent are due to be paid to the U.S. PTO and foreign patent agencies in several stages over the term of the patent. The
U.S. PTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and
other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a
late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment
or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Noncompliance
events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond
to office actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In
such an event, our competitors might be able to enter the market, which would have a material adverse effect on our business.
We may not be able to enforce our intellectual
property rights throughout the world.
Filing, prosecuting and defending
patents on product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights
in some countries outside the United States and Switzerland can be less extensive than those in the United States and Switzerland. In
addition, the laws of some foreign countries do not protect intellectual property to the same extent as laws in the United States and
Switzerland. Consequently, we may not be able to seek to prevent third parties from practicing our inventions in all countries outside
the United States and Switzerland, or from selling or importing products made using our inventions in and into the United States or other
jurisdictions. Competitors, for example, may use our technologies in jurisdictions where we have not obtained patents to develop their
own products and further, may export otherwise infringing products to territories where we have patents, but enforcement is not as strong
as that in the United States and Switzerland.
Many companies have
encountered significant problems in protecting and defending intellectual property in foreign jurisdictions. The legal systems of
certain countries, particularly China and certain other developing countries, do not favor the enforcement of patents, trade secrets
and other intellectual property, particularly those relating to medical devices and biopharmaceutical and biotechnology products,
which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our
proprietary rights generally. To date, we have not sought to enforce any issued patents in these foreign jurisdictions. Proceedings
to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from
other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent
applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits
that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. The requirements for
patentability may differ in certain countries, particularly developing countries. Certain countries in Europe and developing
countries, including China and India, have compulsory licensing laws under which a patent owner may be compelled to grant licenses
to third parties. In those countries, we and our licensors may have limited remedies if patents are infringed or if we or our
licensors are compelled to grant a license to a third party, which could materially diminish the value of those patents. This could
limit our potential revenue opportunities. Accordingly, our efforts to enforce our intellectual property rights around the world may
be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
If we are unable to maintain effective proprietary
rights for our product candidates, we may not be able to compete effectively in our markets.
In addition to the protection
afforded by any patents currently issued, historically, we have relied on trade secret protection and confidentiality agreements to protect
proprietary know-how that is not patentable or that we elect not to patent, processes that are not easily known, knowable or easily ascertainable,
and for which patent infringement is difficult to monitor and enforce and any other elements of our product candidate discovery and development
processes that involve proprietary know-how, information or technology that is not covered by patents. However, trade secrets can be difficult
to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our
employees, consultants, scientific advisors, and contractors. We also seek to preserve the integrity and confidentiality of our data,
trade secrets and intellectual property by maintaining physical security of our premises and physical and electronic security of our information
technology systems. Agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition,
our trade secrets and intellectual property may otherwise become known or be independently discovered by competitors.
We cannot provide any assurances
that our trade secrets and other confidential proprietary information will not be disclosed in violation of our confidentiality agreements
or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information
and techniques. Also, misappropriation or unauthorized and unavoidable disclosure of our trade secrets and intellectual property could
impair our competitive position and may have a material adverse effect on our business. Additionally, if the steps taken to maintain our
trade secrets and intellectual property are deemed inadequate, we may have insufficient recourse against third parties for misappropriating
any trade secret.
Third parties may initiate legal proceedings
alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material
adverse effect on the success of our business.
Our commercial success depends
upon our ability to develop, manufacture, market and sell our product candidates without infringing the proprietary rights of third parties.
There is considerable intellectual property litigation in the pharmaceutical industry. While no such litigation has been brought against
us and we have not been held by any court to have infringed a third party’s intellectual property rights, we cannot guarantee that
our product candidates or use of our product candidates do not infringe third-party patents. It is also possible that we have failed to
identify relevant third-party patents or applications. For example, applications filed before November 29, 2000 and certain applications
filed after that date that will not be filed outside the United States remain confidential until patents issue. Patent applications in
the United States and elsewhere are published approximately 18 months after the earliest filing, which is referred to as the priority
date. Therefore, patent applications covering our product candidates could have been filed by others without our knowledge. Additionally,
pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover
our product candidates.
We may become party to, or
threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our product candidates,
including inter parties review, interference, or derivation proceedings before the U.S. PTO and similar bodies in other countries. Third
parties may assert infringement claims against us based on existing intellectual property rights and intellectual property rights that
may be granted in the future.
If we are found to infringe
a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing
and marketing our product candidates. However, we may not be able to obtain any required license on commercially reasonable terms or at
all. Any inability to secure licenses could result in delays in the introduction of our products or lead to prohibition of the manufacture
or sale of products by us. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access
to the same license. We could be forced, including by court order, to cease commercializing the products subject to the infringing intellectual
property rights. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we
are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates
or force us to cease some of our business operations, which could materially harm our business. Claims that we have misappropriated the
confidential information or trade secrets of third parties could have a similar negative impact on our business.
We may be subject to claims challenging
the inventorship of our patents and other intellectual property.
We may be subject to claims
that former employees, collaborators or other third parties have an interest in our patents or other intellectual property as an inventor
or co-inventor. For example, we may have inventorship disputes arise from conflicting obligations of consultants or others who are involved
in developing our product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship. If
we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such
as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our
business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction
to management and other employees. In addition, we may receive less revenue from future products if any of our employees successfully
claim for compensation for their work in developing our intellectual property, which in turn could impact our future profitability.
Under applicable employment laws, we may
not be able to enforce covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise
of some of our former employees. In addition, employees may be entitled to seek compensation for their inventions irrespective of their
agreements with us.
We generally enter into non-competition
agreements with our employees and certain key consultants. These agreements prohibit our employees and certain key consultants, if they
cease working for us, from competing directly with us or working for our competitors or clients for a limited period of time. We may be
unable to enforce these agreements under the laws of the jurisdictions in which our employees work and it may be difficult for us to restrict
our competitors from benefitting from the expertise our former employees or consultants developed while working for us. Under Swiss law,
a non-compete agreement could be invalidated if, for example, the geographic scope of the non-compete agreement is too broad, or, alternatively,
such an agreement could be deemed by a Swiss court to be an occupation ban. Such actions would make enforcing our non-competition agreements
more challenging and could make it easier for our competitors to employee our previous employees.
In addition, under Swiss law,
if we wish to obtain ownership over inventions developed by our employees, which inventions were developed while performing their employment
activities, but outside the performance of their contractual duties, we are required to compensate the employee for the rights to their
respective inventions. There can be no guarantee that we will be able to obtain any such inventions and the failure to obtain such ownership
rights over employee inventions could have a material adverse effect on our operations and ability to effectively compete.
Risks Related to Our Business Operations
We manage our business through a small number
of key employees and consultants.
As of the date of this annual
report, our key employees and consultants include our Chief Executive Officer, Mr. Alexander Zwyer, our Chief Financial Officer, Nicole
Fernandez–McGovern, and our Chief Scientific Officer, Eric Konofal. Our future growth and success depend on our ability to recruit,
retain, manage and motivate our employees and key consultants. The loss of the services of our Chief Executive Officer or any of our senior
management or the inability to hire or retain experienced management personnel could adversely affect our ability to execute our business
plan and harm our operating results.
In addition, laws and regulations
on executive compensation, including legislation in our home country, Switzerland, may restrict our ability to attract, motivate and retain
the required level of qualified personnel. In Switzerland, legislation affecting public companies has been passed that, among other things,
(i) imposes an annual binding shareholders’ “say-on-pay” vote with respect to the compensation of the members of (a)
the senior management and (b) the board of directors, (ii) prohibits severance, advances, transaction premiums and similar payments to
senior management and directors, and (iii) requires companies to specify various compensation-related matters in their articles of association,
thus requiring them to be approved by a shareholders’ vote.
Because of the specialized
scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific and technical
consultants. In particular, the loss of one or more of our senior management or key consultants could be detrimental to us if we cannot
recruit suitable replacements in a timely manner. We do not currently carry “key person” insurance on the lives of members
of senior management. The competition for qualified personnel in the pharmaceutical industry is intense. Due to this intense competition,
we may be unable to attract and retain qualified personnel necessary for the development of our business or to recruit suitable replacement
personnel.
We may be subject, directly or indirectly,
to federal and state healthcare fraud and abuse laws, false claims laws and health information privacy and security laws. If we are unable
to comply, or have not fully complied, with such laws, we could face substantial penalties.
If we obtain FDA approval
for any of our product candidates and begin commercializing those products in the United States, our operations may be directly or indirectly
through our customers, subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback
Statute, the federal False Claims Act and physician sunshine laws and regulations. These laws may impact, among other things, our proposed
sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and
the states in which we conduct our business. The laws that may affect our ability to operate include:
| |
● |
the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
| |
● |
federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent; |
| |
● |
the Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
| |
● |
HIPAA, as amended by the Health Information Technology and Clinical Health Act, and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; |
| |
● |
the federal physician sunshine requirements under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or the PPACA, requires manufacturers of drugs, devices and medical supplies to report annually to the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians, other healthcare providers and teaching hospitals and ownership and investment interests held by physicians and other healthcare providers and their immediate family members and applicable group purchasing organizations; |
| |
● |
state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; and |
| |
● |
European and other foreign law equivalents of each of the laws, including reporting requirements detailing interactions with and payments to healthcare providers, and the European General Data Protection Regulation, or GDPR, which became effective in May 2018 and contains new provisions specifically directed at the processing of health information, higher sanctions and extra-territoriality measures intended to bring non-EU companies under the regulation, including companies like us that conduct clinical trials in the EU; we anticipate that over time we may expand our business operations to include additional operations in the EU and with such expansion, we would be subject to increased governmental regulation in the EU countries in which we might operate, including the GDPR. |
The scope and enforcement
of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of
the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions
between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements
in the healthcare industry. It is possible that governmental authorities will conclude that our business practices do not comply with
current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If
our operations are found to be in violation of any of these laws or any other related governmental regulations that may apply to us, we
may be subject to significant civil, criminal and administrative penalties, including, without limitation, damages, fines, imprisonment,
disgorgement, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, reputational harm,
additional oversight and reporting obligations if we become subject to a corporate integrity agreement or similar settlement to resolve
allegations of non-compliance with these laws and the curtailment or restructuring of our operations. If any of the physicians or other
healthcare providers or entities with whom we expect to do business is found to be not in compliance with applicable laws, they may be
subject to similar actions, penalties and sanctions. Efforts to ensure that our business arrangements comply with applicable healthcare
laws and regulations, as well as responding to possible investigations by government authorities, can be time- and resource-consuming
and can divert a company’s attention from the business.
Healthcare legislative and regulatory reform
measures may have a material adverse effect on our business and results of operations.
Our industry is highly
regulated and changes in law may adversely impact our business, operations, or financial results. The PPACA was a sweeping measure
intended to, among other things, expand healthcare coverage within the United States, primarily through the imposition of health
insurance mandates on employers and individuals and expansion of the Medicaid program. We anticipate that the PPACA, as well as
other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and an additional
downward pressure on the reimbursement our customers may receive for our products. Further, there have been, and there may continue
to be, judicial and Congressional challenges to certain aspects of the PPACA. For example, the U.S. Tax Cuts and Jobs Act of 2017,
or TCJA, includes a provision repealing the tax-based shared responsibility payment imposed by the PPACA on certain individuals who
fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual
mandate.” Any reduction in reimbursement from Medicare and other government programs may result in a similar reduction in
payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being
able to generate revenue, attain profitability or commercialize our products.
In addition, the delivery
of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of
medicines, is almost exclusively a matter for national, rather than EU, law and policy. National governments and health service providers
have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context.
In general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on the pricing and reimbursement
of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory burdens on those wishing to
develop and market products, this could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval
activities and affect our ability to commercialize any products for which we obtain marketing approval.
We are currently unable to
predict what additional legislation or regulation, if any, relating to the healthcare industry may be enacted in the future or what effect
recently enacted federal legislation or any such additional legislation or regulation would have on our business. The pendency or approval
of such proposals or reforms could result in a decrease in the price of our common shares or Warrants or limit our ability to raise capital
or to enter into collaboration agreements for the further development and potential commercialization of our products.
The use of any of our product candidates
could result in product liability or similar claims that could be expensive, damage our reputation and harm our business.
Our business exposes us to
an inherent risk of potential product liability or similar claims. The pharmaceutical industry has historically been litigious, and we
face financial exposure to product liability or similar claims if the use of any of our products were to cause or contribute to injury
or death. There is also the possibility that defects in the design or manufacture of any of our products might necessitate a product recall.
Although we plan to maintain product liability insurance, the coverage limits of these policies may not be adequate to cover future claims.
In the future, we may be unable to maintain product liability insurance on acceptable terms or at reasonable costs and such insurance
may not provide us with adequate coverage against potential liabilities. A product liability claim, regardless of merit or ultimate outcome,
or any product recall could result in substantial costs to us, damage to our reputation, customer dissatisfaction and frustration and
a substantial diversion of management attention. A successful claim brought against us in excess of, or outside of, our insurance coverage
could have a material adverse effect on our business, financial condition and results of operations.
Risks Related to the Ownership of Our Securities
The market price of our common shares and
Warrants may be highly volatile, and you may not be able to resell your shares or Warrants at or above the initial public offering price.
The trading price of each
of our common shares and Warrants is likely to be volatile. The following factors, in addition to other risk factors described in this
section, may have a significant impact on the market price of such securities:
| |
● |
unsatisfactory results of clinical trials; |
| |
|
|
| |
● |
announcements of regulatory approval or the failure to obtain it, or specific label indications or patient populations for its use, or changes or delays in the regulatory review process; |
| |
|
|
| |
● |
announcements of therapeutic innovations or new products by us or our competitors; |
| |
|
|
| |
● |
adverse actions taken by regulatory authorities with respect to our clinical trials, manufacturing supply chain or sales and marketing activities; |
| |
|
|
| |
● |
changes or developments in laws or regulations applicable to the treatment of the symptoms of narcolepsy, ADHD, or any other indication that we may seek to develop, or the use of mazindol to treat any such indications; |
| |
|
|
| |
● |
any adverse changes to our relationship with manufacturers or suppliers; |
| |
|
|
| |
● |
any intellectual property infringement actions in which we may become involved; |
| |
|
|
| |
● |
announcements concerning our competitors or the pharmaceutical industry in general; |
| |
|
|
| |
● |
achievement of expected product sales and profitability or our failure to meet expectations; |
| |
|
|
| |
● |
our commencement of, or involvement in, litigation; |
| |
|
|
| |
● |
any major changes in our board of directors or management; |
| |
|
|
| |
● |
our ability to recruit and retain qualified regulatory, research and development personnel; |
| |
|
|
| |
● |
legislation in the United States relating to the sale or pricing of pharmaceuticals; |
| |
|
|
| |
● |
the depth of the trading market in our common shares and Warrants; |
| |
|
|
| |
● |
economic weakness, including rising or sustained high interest rates and high inflation, or political instability in particular foreign economies and markets; |
| |
|
|
| |
● |
business interruptions resulting from a local or worldwide pandemic (such as the COVID-19 pandemic), geopolitical instability (such as the war in Ukraine and Israel’s multi-front war), and other conditions beyond our control; |
| |
|
|
| |
● |
the granting or exercise of employee stock options or other equity awards; and |
| |
|
|
| |
● |
changes in investors’ and securities analysts’ perception of the business risks and conditions of our business. |
In addition, the stock
market in general, and Nasdaq in particular, have experienced extreme price and volume fluctuations that have often been unrelated
or disproportionate to the operating performance of small companies. For example, during 2023, the sale prices of our common shares
ranged from a high of $75.2 per share to a low of $10.4 per share and during 2024, the sale prices of our common shares ranged from
a high of $23.52 per share to a low of $1.82 per share. During this time, we do not believe that we have experienced any material
changes in our financial condition or results of operations that would explain such price volatility or trading volume; however, we
have sold equity which was dilutive to existing shareholders. These broad market fluctuations may adversely affect the trading price
of our securities. Additionally, these and other external factors have caused and may continue to cause the market price and demand
for our common shares to fluctuate substantially, which may limit or prevent our shareholders from readily selling their shares and
may otherwise negatively affect the liquidity of our shares.
We have identified material weaknesses in
our internal control over financial reporting. If our remediations are not effective, or if we experience additional material weaknesses
in the future or otherwise fail to maintain an effective system of internal controls in the future, we may not be able to accurately or
timely report our financial conditions or results or operations, or prevent fraud, which may adversely affect investor confidence in our
Company and as a result, the market price of our common shares and Warrants.
As a public company, we are
required to maintain internal control over financial reporting and will be required to report any material weaknesses in such internal
control. Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, requires that we include a report from management on
the effectiveness of our internal control over financial reporting in our annual report on Form 20-F. In addition, once we cease to be
an EGC as such term is defined in the JOBS Act, our independent registered public accounting firm must attest to and report on the effectiveness
of our internal control over financial reporting. According to the U.S. Public Company Accounting Oversight Board, a material weakness
is a deficiency or combination of deficiencies in internal control over financial reporting such that there is a reasonable possibility
that a material misstatement of our financial statements will not be prevented or detected on a timely basis. If we fail to remediate
the material weaknesses, or are otherwise unable to maintain effective internal control over financial reporting, management could be
required to expend significant resources and we could fail to meet our public reporting requirements on a timely basis, and be subject
to fines, penalties, investigations or judgements, all of which could negatively affect investor confidence and adversely impact our stock
price.
As of December 31, 2024, we
have material weaknesses in our internal control over financial reporting, relating to a lack of sufficient number of trained professionals
with an appropriate level of accounting knowledge to design and maintain controls over the preparation of financial statements, and relating
to a lack of maintaining appropriate segregation of duties.
During the year ended December
31, 2024, we have taken steps to remediate the material weaknesses and to strengthen our internal control over financial reporting. We
conducted an analysis to identify the underlying reasons for the material weaknesses. This involved assessing risk factors, understanding
process gaps, and pinpointing areas that needed improvement. As part of our remediation, we have transitioned the accounting processes
to a dedicated team with U.S. GAAP and SEC reporting knowledge, with the support of additional external advisors. While we believe that
these efforts will improve our internal control over financial reporting in accordance with U.S. GAAP and SEC reporting requirements,
the implementation of these measures is ongoing and will require validation and testing of the design and operating effectiveness of internal
controls over a sustained period of financial reporting cycles. We cannot assure you that the measures we have taken to date, and are
continuing to implement, will be sufficient to establish and maintain effective internal control over financial reporting.
The Warrants we have issued are speculative
in nature.
The Warrants we have offered
and sold do not confer any rights of common share ownership on their holders, such as voting rights or the right to receive dividends,
but rather merely represent the right to acquire common shares at a fixed price for a limited period of time. Specifically, holders of
the Warrants we have issued may exercise their right to acquire the common shares and pay an exercise price per common share as contemplated
in such holder’s Warrant, prior to five years from the date of issuance, after which date any unexercised Warrants will expire and
have no further value.
If we were to be characterized as a “passive
foreign investment company” for U.S. tax purposes, U.S. holders of our common shares and Warrants could have adverse U.S. income
tax consequences.
In general, we will be treated
as a passive foreign investment company, or a PFIC, for U.S. federal income tax purposes in any taxable year in which either (1) at least
75% of our gross income is “passive income” or (2) on average at least 50% of our assets by value produce passive income or
are held for the production of passive income. Passive income for this purpose generally includes, among other things, certain dividends,
interest, royalties, rents and gains from commodities and securities transactions and from the sale or exchange of property that gives
rise to passive income. Passive income also includes amounts derived by reason of the temporary investment of funds, including those raised
in a public offering. In determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each
corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account. We do not believe that
we will be deemed a PFIC for 2024. If we are a PFIC in any taxable year during which a U.S. taxpayer holds the common shares and Warrants,
such U.S. taxpayer would be subject to certain adverse U.S. federal income tax rules. In particular, except with respect to the Warrants
(unless exercised), if the U.S. taxpayer did not make an election to treat us as a “qualified electing fund,” or QEF, or make
a “mark-to-market” election, then “excess distributions” to the U.S. taxpayer, and any gain realized on the sale
or other disposition of the common shares by the U.S. taxpayer: (1) would be allocated ratably over the U.S. taxpayer’s holding
period for the common shares; (2) the amount allocated to the current taxable year and any period prior to the first day of the first
taxable year in which we were a PFIC would be taxed as ordinary income; and (3) the amount allocated to each of the other taxable years
would be subject to tax at the highest rate of tax in effect for the applicable class of taxpayer for that year, and an interest charge
for the deemed deferral benefit would be imposed with respect to the resulting tax attributable to each such other taxable year. In addition,
if the U.S. Internal Revenue Service, or the IRS, determines that we are a PFIC for a year with respect to which we have determined that
we were not a PFIC, it may be too late for a U.S. taxpayer to make a timely QEF or mark-to-market election. U.S. taxpayers that have held
the common shares during a period when we were a PFIC will be subject to the foregoing rules, even if we cease to be a PFIC in subsequent
years, subject to exceptions for U.S. taxpayer who made a timely QEF or mark-to-market election. A U.S. taxpayer can make a QEF election
by completing the relevant portions of and filing IRS Form 8621 in accordance with the instructions thereto. We intend to make available
to U.S. taxpayers upon request the information needed in order to complete IRS Form 8621 and to make and maintain a valid QEF election
for any year in which we or any of our subsidiaries are a PFIC. U.S. taxpayers that hold the common shares are strongly urged to consult
their tax advisors about the PFIC rules, including tax return filing requirements and the eligibility, manner, and consequences to them
of making a QEF or mark-to-market election with respect to the common shares in the event that we are a PFIC. See Item 10.E. “Taxation-U.S.
Federal Income Tax Consequences-Passive Foreign Investment Companies” for additional information.
We have not paid, and do not intend to pay,
dividends on our common shares and, therefore, unless our traded securities appreciate in value, our investors may not benefit from holding
our common shares.
We have never declared or
paid cash dividends on our common shares. We do not anticipate paying any cash dividends on our common shares in the foreseeable future.
Moreover, Swiss Law imposes certain restrictions on our ability to declare and pay dividends. See Item 8.A. “Dividends” for
additional information.
Our failure to meet the continued listing requirements Nasdaq
could result in a delisting of our securities or us seeking to transfer our listed securities to a different exchange.
On January 9, 2024, we
received a letter from the staff (the “Staff”) of the Listing Qualifications Department of Nasdaq indicating that we did
not meet the minimum of $2.5 million in shareholders’ equity required for continued listing on Nasdaq pursuant to Nasdaq
Listing Rule 5550(b)(1) or the alternative compliance standards relating to the market value of listed securities or net income from
continuing operations, or the Minimum Stockholders’ Equity Requirement. The letter also indicated that we would be provided
with a period of 45 calendar days to submit a plan to regain compliance, and if such plan is accepted by Nasdaq, we may be granted
up to 180 calendar days from January 9, 2024 in which to regain compliance. On February 23, 2024, we submitted our plan, on time, to
regain compliance with Nasdaq Listing Rule 5550(b)(1). On March 7, 2024, the Staff notified us that Nasdaq had accepted the plan
submitted and the Company has been afforded an additional 180 day calendar days from the date of the letter, or until July 8, 2024,
to evidence compliance.
On April 17, 2024, we received
a determination letter from the Staff that, unless we timely request a hearing before a Nasdaq Hearings Panel (the “Panel”),
our securities would be subject to delisting due to our failure to maintain at least a $1.00 bid price per share over the course of 30
consecutive business days pursuant to Nasdaq Listing Rule 5550(a)(2). In addition, on May 22, 2024, we also received an additional determination
letter from the Staff notifying us that we did not comply with the minimum $2,500,000 stockholders’ equity requirement for continued
listing set forth in Nasdaq Listing Rule 5550(b), and that the additional delinquency may serve as a separate basis for the delisting
of our securities from Nasdaq. We requested a hearing with the Panel, and on June 4, 2024, we presented our compliance plan to the Panel.
The Panel granted us an extension until October 14, 2024, to regain compliance with Nasdaq Listing Rule 5550(a)(2) and Nasdaq Rule
5550(b)(1), subject to us meeting certain financial, share price and other requirements by September 3, 2024 and October 14, 2024.
On September 27, 2024, we
effectuated a 1-for-40 reverse share split in order to regain compliance with Nasdaq Listing Rule 5550(a)(2). On September 27, 2024, the
Panel reconfirmed to us that we had until October 14, 2024 to regain compliance with Nasdaq Listing Rule 5550(a)(2) and 5550(b)(1).
On October 28, 2024, we received notice from the Staff informing us that we regained full compliance with the bid price requirement in
Listing Rule 5550(a)(2) and the equity requirement in Listing Rule 5550(b)(1).
We have in the past, and may
in the future, be unable to comply with certain of the listing standards that we are required to meet to maintain the listing of our common
shares on Nasdaq. If we fail to satisfy the continued listing requirements of Nasdaq, such as minimum stockholders’ equity requirements
or minimum bid price requirements, Nasdaq may take steps to delist our common shares. Such a delisting would have a negative effect on
the price of our common shares, impair the ability to sell or purchase our common shares when persons wish to do so, and any delisting
materially adversely affect our ability to raise capital or pursue strategic restructuring, refinancing or other transactions on acceptable
terms, or at all. Delisting from Nasdaq could also have other negative results, including the potential loss of institutional investor
interest and fewer business development opportunities, as well as a limited amount of news and analyst coverage of us. Delisting could
also result in a determination that our common shares are a “penny stock,” which would require brokers trading in our common
shares to adhere to more stringent rules, possibly resulting in a reduced level of trading activity in the secondary market for our common
shares. In the event of a delisting, we would attempt to take actions to restore our compliance with Nasdaq’s listing requirements,
but we can provide no assurance that any such action taken by us would allow our securities to become listed again, stabilize the market
price or improve the liquidity of our securities, prevent our common shares from dropping below the Nasdaq minimum bid price requirement
or prevent future non-compliance with Nasdaq’s listing requirements.
The JOBS Act allows us to postpone the date
by which we must comply with some of the laws and regulations intended to protect investors and to reduce the amount of information we
provide in our reports filed with the SEC, which could undermine investor confidence in our Company and adversely affect the market price
of our common shares and Warrants.
For so long as we remain an
EGC, we intend to take advantage of certain exemptions from various requirements that are applicable to public companies that are not
EGCs including:
| |
● |
the provisions of the Sarbanes-Oxley Act requiring that our independent registered public accounting firm provide an attestation report on the effectiveness of our internal control over financial reporting; |
| |
● |
Section 107 of the JOBS Act, which provides that an EGC can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, or the Securities Act, for complying with new or revised accounting standards. This means that an EGC can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We are electing to delay such adoption of new or revised accounting standards. As a result of this adoption, our financial statements may not be comparable to companies that comply with the public company effective date; and |
| |
● |
any rules that may be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotation or a supplement to the auditor’s report on the financial statements. |
We intend to take advantage
of these exemptions until we are no longer an EGC. We will remain an EGC until the earlier of (1) the last day of the fiscal year (a)
following the fifth anniversary of the date of the completion of our initial public offering, (b) in which we have total annual gross
revenue of at least $1.235 billion, or (c) in which we are deemed to be a large accelerated filer, as defined in the rule under the Exchange
Act, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
We cannot predict if investors
will find our common shares and Warrants less attractive because we may rely on these exemptions. If some investors find our common shares
and Warrants less attractive as a result, there may be a less active trading market for each such traded security, and the trading price
of each may be more volatile and may decline.
The requirements associated with being a
public company require significant company resources and management attention.
We are subject to the reporting
requirements of the Exchange Act, Nasdaq listing requirements and other applicable securities rules and regulations. The Exchange Act
requires that we file periodic reports with respect to our business and financial condition and maintain effective disclosure controls
and procedures and internal control over financial reporting. In addition, subsequent rules implemented by the SEC and Nasdaq may also
impose various additional requirements on public companies. As a result, we will incur additional legal, accounting and other expenses,
particularly after we are no longer an EGC as defined in the JOBS Act. We estimate that these expenses will be in excess of one million
dollars annually. We have made changes to our corporate governance standards, disclosure controls and financial reporting and accounting
systems to meet our reporting obligations. The measures we take, however, may not be sufficient to satisfy our obligations as a public
company, which could subject us to delisting of our common shares, fines, sanctions and other regulatory action and potentially civil
litigation.
After we are no longer an
EGC, we expect to incur significant expenses and devote substantial management efforts toward ensuring compliance with the requirements
of Section 404 of the Sarbanes-Oxley Act and the other rules and regulations of the SEC.
Risks Related to Swiss Law and Our Operations
in Switzerland
We are a Swiss corporation. The rights of
our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions.
We are a Swiss
corporation. Our corporate affairs are governed by our amended articles of association and by the laws governing companies,
including listed companies, incorporated in Switzerland. The rights of our shareholders and the responsibilities of members of our
board of directors may be different from the rights and obligations of shareholders and directors of companies governed by the laws
of U.S. jurisdictions. In the performance of its duties, our board of directors is required by Swiss law to consider the interests
of our Company, our shareholders, our employees and other stakeholders, in all cases with due observation of the principles of
reasonableness and fairness. It is possible that some of these parties will have interests that are different from, or in addition
to, your interests as a shareholder. Swiss corporate law limits the ability of our shareholders to challenge resolutions made or
other actions taken by our board of directors in court. Our shareholders generally are not permitted to file a suit to reverse a
decision or an action taken by our board of directors but are instead only permitted to seek damages for breaches of fiduciary duty.
However, if a board resolution has been invalidly formed, anyone, including the shareholders, may claim that such an invalid
resolution shall be declared null and void. As a matter of Swiss law, shareholder claims against a member of our board of directors
for breach of fiduciary duty would have to be brought to the competent court at the legal seat of the Company, or where the relevant
member of our board of directors is domiciled. In addition, under Swiss law, any claims by our shareholders against us must be
brought to the competent court at the legal seat of the Company.
Our common shares are issued under the laws
of Switzerland, which may not protect investors in a similar fashion afforded by incorporation in a U.S. state.
We are organized under the
laws of Switzerland. However, there can be no assurance that Swiss law will not change in the future or that it will serve to protect
investors in a similar fashion afforded under corporate law principles in the United States, which could adversely affect the rights of
investors.
Our status as a Swiss corporation may limit
our flexibility with respect to certain aspects of capital management and may cause us to be unable to make distributions without subjecting
our shareholders to Swiss withholding tax.
The amended Swiss corporate
law, effective January 1, 2023, allows our shareholders to implement a so called capital band (Kapitalband) in the articles of
association which authorizes the board of directors to increase and/or decrease the share capital within a certain bandwidth without additional
shareholder approval. This authorization is limited to 50% of the existing registered share capital and the authorization requires renewal
by the shareholders every five years or at its earlier expiration. Additionally, subject to specified exceptions, Swiss law grants pre-emptive
subscription rights to existing shareholders to subscribe to any new issuance of shares. Swiss law also does not provide as much flexibility
in the various terms that can attach to different classes of shares as the laws of some other jurisdictions. Swiss law also reserves for
approval by shareholders certain corporate actions over which a board of directors would have authority in some other jurisdictions. For
example, dividends must be approved by shareholders. These Swiss law requirements relating to our capital management may limit our flexibility,
and situations may arise where greater flexibility would have provided substantial benefits to our shareholders.
Under Swiss law, a Swiss corporation may pay dividends only if the
corporation has sufficient distributable profits from previous fiscal years, or if the corporation has distributable reserves, each as
evidenced by its audited statutory balance sheet. Furthermore, a Swiss corporation may pay interim dividends (“Zwischendividende”)
if the corporation has sufficient distributable profits during the current fiscal year. The remaining provisions applicable to the dividends
are also applicable to the interim dividends. Freely distributable reserves are generally booked either as “free reserves”
or as “capital contributions” (contributions received from shareholders) in the “reserve from capital contributions.”
Distributions may be made out of issued share capital - the aggregate nominal value of a company’s issued shares - only by way of
a capital reduction. As of December 31, 2024, we had CHF 56,373,233 (approximately $60,408,490) of qualifying capital contributions and
CHF 2,652,999.2 (approximately $2,909,013.62) of registered share capital (consisting of 1,703,345 common shares each with a nominal value
of CHF 0.80, 598,539 preferred shares each with a nominal value of CHF 0.80 and 1,014,365 preferred participation certificates each with
a nominal value of CHF 0.80) on our audited statutory balance sheet (which audit was performed by our independent registered public accounting
firm), as required pursuant to Swiss law. We will not be able to pay dividends or make other distributions to shareholders on a Swiss
withholding tax-free basis in excess of that amount unless we increase our share capital or our reserves from capital contributions. We
would also be able to pay dividends out of distributable profits or freely distributable reserves but such dividends would be subject
to Swiss withholding taxes. There can be no assurance that we will have sufficient distributable profits, free reserves, reserves from
capital contributions or registered share capital to pay a dividend or effect a capital reduction, that our shareholders will approve
dividends or capital reductions proposed by us, or that we will be able to meet the other legal requirements for dividend payments or
distributions as a result of capital reductions.
Generally, Swiss withholding
tax of 35% is due on dividends and similar distributions to our shareholders, regardless of the place of residency of the shareholder,
unless the distribution is made to shareholders out of (i) a reduction of nominal value or (ii) assuming certain conditions are met, qualifying
capital contributions accumulated on or after January 1, 1997. A U.S. holder that qualifies for benefits under the Convention between
United States of America and Switzerland for the Avoidance of Double Taxation with Respect to Taxes on Income, which we refer to as the
“U.S.-Swiss Treaty,” may apply for a refund of the tax withheld in excess of the 15% treaty rate (or in excess of the 5% reduced
treaty rate for qualifying corporate shareholders with at least 10% participation in our voting shares, or for a full refund in the case
of qualified pension funds).
There can be no assurance
that we will have sufficient qualifying capital contributions to pay dividends free from Swiss withholding tax, or that Swiss withholding
rules will not be changed in the future. In addition, we cannot provide assurance that the current Swiss law with respect to distributions
out of qualifying capital contributions will not be changed or that a change in Swiss law will not adversely affect us or our shareholders,
in particular as a result of distributions out of qualifying capital contributions becoming subject to additional corporate law or other
restrictions. In addition, over the long term, the amount of par value available to us for nominal value reductions or qualifying capital
contributions available to us to pay out as distributions is limited. If we are unable to make a distribution through a reduction in nominal
value or out of qualifying capital contributions, we may not be able to make distributions without subjecting our shareholders to Swiss
withholding taxes.
Our securities are not listed in Switzerland,
our home jurisdiction. As a result, our shareholders will not benefit from certain provisions of Swiss law that are designed to protect
shareholders in a public takeover offer or a change-of-control transaction and may not be protected in the same degree in a public takeover
offer or a change-of-control transaction as are shareholders of certain U.S. companies or in a Swiss company listed in Switzerland.
Because our securities are
listed exclusively on Nasdaq and not in Switzerland, our shareholders will not benefit from the protection afforded by certain provisions
of Swiss law that are designed to protect shareholders in the event of a public takeover offer or a change-of-control transaction. For
example, Article 120 of the Swiss Financial Market Infrastructure Act and its implementing provisions require investors to disclose their
interest in our company if they reach, exceed or fall below certain ownership thresholds. Similarly, the Swiss takeover regime imposes
a duty on any person or group of persons who acquires more than one-third of a company’s voting rights to make a mandatory offer
for all of the company’s outstanding listed equity securities. In addition, the Swiss takeover regime imposes certain restrictions
and obligations on bidders in a voluntary public takeover offer that are designed to protect shareholders. However, these protections
are applicable only to issuers that list their equity securities in Switzerland and, because our securities are listed exclusively on
Nasdaq, will not be applicable to us. Furthermore, since Swiss law restricts our ability to implement rights plans or U.S.-style “poison
pills,” our ability to resist an unsolicited takeover attempt or to protect minority shareholders in the event of a change of control
transaction may be limited. Therefore, our shareholders may not be protected in the same degree in a public takeover offer or a change-of-control
transaction as are shareholders in certain U.S. companies or in a Swiss company listed in Switzerland.
U.S. shareholders may not be able to obtain
judgments or enforce civil liabilities against us or our senior management or members of our board of directors.
We are organized under
the laws of Switzerland and our registered office and domicile is located in Kloten (Zurich), Switzerland. Moreover, a majority of
our directors and senior management are not residents of the United States, and all or a substantial portion of the assets of such
persons are located outside the United States. As a result, it may not be possible for investors to effect service of process within
the United States upon us or upon such persons or to enforce against them judgments obtained in U.S. courts, including judgments in
actions predicated upon the civil liability provisions of the federal securities laws of the United States. We have been advised by
our Swiss counsel that there is doubt as to the enforceability in Switzerland of original actions, or in actions for enforcement of
judgments of U.S. courts, of civil liabilities to the extent solely predicated upon the federal and state securities laws of the
United States. Original actions against persons in Switzerland based solely upon the U.S. federal or state securities laws are
governed, among other things, by the principles set forth in the Swiss Federal Act on Private International Law. This statute
provides that the application of provisions of non-Swiss law by the courts in Switzerland shall be precluded if the result is
incompatible with Swiss public policy. Also, certain mandatory provisions of Swiss law may be applicable regardless of any other law
that would otherwise apply.
Switzerland and the United
States do not have a treaty providing for reciprocal recognition and enforcement of judgments in civil and commercial matters. The recognition
and enforcement of a judgment of the courts of the United States in Switzerland is governed by the principles set forth in the Swiss Federal
Act on Private International Law. This statute provides in principle that a judgment rendered by a non-Swiss court may be recognized in
Switzerland only if:
| |
● |
the non-Swiss court had jurisdiction pursuant to the Swiss Federal Act on Private International Law; |
| |
● |
the judgment of such non-Swiss court has become final and non-appealable; |
| |
● |
the judgment does not contravene Swiss public policy; |
| |
● |
the court procedures and the service of documents leading to the judgment were in accordance with the due process of law; and |
| |
● |
no proceeding involving the same position and the same subject matter was first brought in Switzerland, or adjudicated in Switzerland, or was earlier adjudicated in a third state and this decision is recognizable in Switzerland. |
Exchange rate fluctuations between the U.S.
dollar and the Swiss Franc may negatively affect our results.
Under our existing agreements,
we make a significant number of payments in U.S. dollars, Swiss Francs and Euro. Since our reporting currency is the U.S. dollar, financial
line items are converted into U.S. dollars at the applicable exchange rates. As a result, changes and fluctuations in currency exchange
rates between the Swiss Franc, Euro the U.S. dollar could have a material adverse effect on our operating results.
General Risk Factors
We are a foreign private issuer within the
meaning of the rules under the Exchange Act, and as such we are exempt from certain provisions applicable to U.S. domestic public companies,
which may result in less protection than is accorded to investors under rules applicable to domestic U.S. issuers.
Our status as a foreign private
issuer exempts us from compliance with certain SEC laws and regulations and certain regulations of Nasdaq, including the proxy rules,
the short-swing profits recapture rules, and certain governance requirements such as independent director oversight of the nomination
of directors and executive compensation. In addition, we are not required under the Exchange Act to file current reports and financial
statements with the SEC as frequently or as promptly as U.S. domestic companies whose securities are registered under the Exchange Act
and we are exempt from filing quarterly reports with the SEC. Furthermore, as a foreign private issuer, we are also not subject to the
requirements of Regulation FD (Fair Disclosure) promulgated under the Exchange Act.
These exemptions and leniencies
will reduce the frequency and scope of information and protections to which you are entitled as an investor.
We are required to file an
annual report on Form 20-F within four months of the end of each fiscal year. In addition, we intend to publish our results on a half-yearly
basis as press releases, distributed pursuant to the rules and regulations of the Nasdaq. Press releases relating to financial results
and material events will also be furnished to the SEC on Form 6-K. However, the information we are required to file with or furnish to
the SEC will be less extensive and less timely compared to that required to be filed with the SEC by U.S. domestic issuers. As a result,
you may not be afforded the same protections or information that would be made available to you were you investing in a U.S. domestic
issuer.
Raising additional capital would cause dilution
to our existing shareholders, and may affect the rights of existing shareholders.
In future, we may seek additional
capital through a combination of private and public equity offerings, debt financings and collaborations and strategic and licensing arrangements.
To the extent that we raise additional capital through the issuance of equity or convertible debt securities, your ownership interest
will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a shareholder. Future
sales of our common shares or of securities convertible into our common shares, or the perception that such sales may occur, could cause
immediate dilution and adversely affect the market price of our common shares and the Warrants.
Our business is subject to a variety of
U.S. and international laws and regulations, including those regarding privacy, cybersecurity and data protection. Any failure of our
platform to comply with applicable laws and regulations could harm our business, operating results and financial condition.
Our operations may from time
to time involve cross-border transfer of data, which may subject us and our customers that use our products to privacy, cybersecurity
and data protection-related laws and regulations that impose obligations in connection with the collection, processing and use of personal
data, financial data, health or other similar data and general cybersecurity. Multiple jurisdictions have adopted or proposed limitations
on, or requirements regarding, the collection, distribution, use, security and storage of information, including personally identifiable
information of individuals.
In the United States, the
U.S. Federal Trade Commission and numerous state attorneys general are applying federal and state consumer protection laws to impose standards
on the online collection, use and dissemination of data, and to the security measures applied to such data. We continue to see increased
regulation of privacy cybersecurity and data protection, including the adoption of more stringent subject matter specific state laws in
the United States. For example, in 2018, California enacted the California Consumer Privacy Act, or the CCPA, which took effect on January 1,
2020. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal
information sharing, and receive detailed information about how their personal information is used. The CCPA provides for civil penalties
for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA may
increase our compliance costs and potential liability, and we may be required to modify our practices and take additional steps in an
effort to comply with the CCPA. Some observers have noted that the CCPA could mark the beginning of a trend toward more stringent state
privacy legislation in the United States, which could increase our potential liability and adversely affect our business.
Similarly, many other
countries and governmental bodies, including the EU member states, have laws and regulations concerning the collection and use of
personal data obtained from individuals located in the EU or by businesses operating within their jurisdiction, which are often more
restrictive than those in the United States. Laws and regulations in these jurisdictions apply broadly to the collection, use,
storage, disclosure and security of personal data that identifies or may be used to identify an individual, such as names, telephone
numbers, email addresses and, in certain circumstances, IP addresses and other online identifiers. For example, the EU has adopted
the GDPR, which enhances data protection obligations for businesses and requires service providers (data processors) processing
personal data on behalf of customers to cooperate with European data protection authorities, implement security measures and keep
records of personal data processing activities. The GDPR also extends the geographical scope of EU data protection law to non-EU
entities under certain conditions, tightens existing EU data protection principles and creates new obligations for companies and new
rights for individuals. Noncompliance with the GDPR can trigger fines equal to or greater of €20 million or 4% of global
annual revenues. Given the breadth and depth of its obligations, working to meet the requirements of the GDPR has required
significant time and resources, including a review of our technology and systems currently in use against the requirements of the
GDPR. There are also additional EU laws and regulations (and member states implementations thereof) which govern the protection of
consumers and of electronic communications. We have taken measures to address certain obligations under the GDPR and to make us GDPR
compliant, but we may be required to take additional steps in order to comply with the GDPR. If our efforts to comply with GDPR or
other applicable EU laws and regulations are not successful, we may be subject to penalties and fines that would adversely impact
our business and operating results, and our ability to conduct business in the EU could be significantly impaired.
We also continue to see jurisdictions
imposing data localization laws, which require personal information, or certain subcategories of personal information to be stored in
the jurisdiction of origin. These regulations may inhibit our ability to expand into those markets or prohibit us from continuing to offer
services in those markets without significant additional costs.
Additionally, although we
endeavor to have our products and platform comply with applicable laws and regulations, these and other obligations may be modified, they
may be interpreted and applied in an inconsistent manner from one jurisdiction to another, and they may conflict with one another, other
regulatory requirements, contractual commitments or our practices. We expect that there will continue to be new proposed laws, rules of
self- regulatory bodies, regulations and industry standards concerning privacy, data protection and information security in the United
States, the EU and other jurisdictions, and we cannot yet determine the impact such future laws, rules, regulations and standards may
have on our business. The uncertainty and changes in the requirements of multiple jurisdictions may increase the cost of compliance, delay
or reduce demand for our services, restrict our ability to offer services in certain locations, impact our customers’ ability to
deploy our products and solutions in certain jurisdictions, or subject us to claims and litigation from private actors and investigations,
proceedings, and sanctions by data protection regulators, all of which could harm our business, financial condition and operating results.
We also may be bound by contractual
obligations relating to our collection, use and disclosure of personal, financial and other data or may find it necessary or desirable
to join industry or other self-regulatory bodies or other privacy, cybersecurity or data protection-related organizations that require
compliance with their rules pertaining to privacy and data protection.
Any failure or perceived failure
by us, our products or our platform to comply with new or existing U.S., EU or other foreign privacy, cybersecurity or data protection
laws, regulations, policies, industry standards or legal obligations, any failure to bind our suppliers and contractors to appropriate
agreements or to manage their practices or any systems failure or security incident that results in the unauthorized access to, or acquisition,
release or transfer of, personally identifiable information or other data relating to customers or individuals may result in governmental
investigations, inquiries, enforcement actions and prosecutions, private claims and litigation, fines and penalties, adverse publicity
or potential loss of business.
We may be subject to securities litigation,
which is expensive and could divert management attention.
In the past, companies that
have experienced volatility in the market price of their shares have been subject to securities class action litigation. We may be the
target of this type of litigation in the future. Litigation of this type could result in substantial costs and diversion of management’s
attention and resources, which could seriously hurt our business. Any adverse determination in litigation could also subject us to significant
liabilities.
Sales of a substantial number of shares
of our common shares in the public market by our existing shareholders could cause our share price to fall.
Sales of a substantial
number of our common shares in the public market or the perception that these sales might occur, could depress the market price of
our common shares and could impair our ability to raise capital through the sale of additional equity securities. We are unable to
predict the effect that sales may have on the prevailing market price of our common shares. In addition, shares held by our
shareholders prior to our initial public offering may be available for re-sale as the lock up period set forth in certain lock-up
agreements executed by certain company insiders relating to our initial public offering has expired. In addition, shares issued or
issuable upon exercise of vested options and exercised Warrants as of the expiration of the lock-up period are eligible for sale.
Sales of shares by these shareholders could have a material adverse effect on the trading price of our common shares.
If securities or industry analysts do not
publish or cease publishing research reports about us, our business or our market, or if they adversely change their recommendations or
publish negative reports regarding our business or our shares and Warrants, the trading prices of our shares and Warrants and trading
volume of our shares and Warrants could decline.
The trading market for our
common shares and Warrants will be influenced by the research reports that industry or securities analysts may publish about us, our business,
our market or our competitors. We do not have any control over these analysts and we cannot provide any assurance that analysts will cover
us or provide favorable coverage. If any of the analysts who may cover us adversely change their recommendation regarding our shares and
Warrants, or provide more favorable relative recommendations about our competitors, the trading price of our shares and Warrants would
likely decline. If any analyst who may cover us were to cease coverage of our Company or fail to regularly publish reports on us, we could
lose visibility in the financial markets, which in turn could cause the trading price of our shares and Warrants and trading volume of
our shares and Warrants to decline.
Risks Related to the Merger
If NLS cannot satisfy, or continue to satisfy, the initial listing requirements and
other rules of Nasdaq, NLS’s securities may not be listed or may be delisted, which could negatively impact the price of its securities
and your ability to sell them.
Under the Merger Agreement,
each party’s obligation to complete the Merger is subject to the satisfaction or waiver of various conditions, including common
shares shall remain listed on the Nasdaq and shall not be subject to a delisting notice from Nasdaq. The notification form for the listing
of common shares to be issued in connection with the Merger shall have been accepted and approved (subject to official notice of issuance),
and the Nasdaq listing application for initial listing of NLS following the Merger and of common shares being issued pursuant to the Merger
Agreement shall have been approved.
NLS is currently seeking to
have its securities approved for listing on Nasdaq in connection with the Merger. NLS cannot assure you that it will be able to meet those
initial listing requirements at the time of the Closing. Even if NLS’s securities are listed on Nasdaq, it cannot assure you that
its securities will continue to be listed on Nasdaq. In addition, following the Merger, to maintain its listing on Nasdaq, NLS will be
required to comply with certain continued listing standards of Nasdaq including those regarding minimum shareholders’ equity, minimum
share price, minimum market value of publicly held shares, minimum number of shareholders and various additional requirements. Even if
NLS initially meets the listing requirements and other applicable rules of Nasdaq, NLS may not be able to continue to satisfy these requirements
and applicable rules. If NLS is unable to satisfy Nasdaq criteria for maintaining its listing, its securities could be subject to delisting.
If Nasdaq does not list NLS’s
securities, or subsequently delists its securities from trading, NLS could face significant consequences, including:
| ● | failure to satisfy a closing condition to the Merger and the
inability to therefore complete the Merger; |
| ● | a limited availability for market quotations for its securities; |
| ● | reduced liquidity with respect to its securities; |
| ● | a determination that its Common Shares are “penny stock,”
which will require brokers trading in its Common Shares to adhere to more stringent rules and possibly result in a reduced level of trading
activity in the secondary trading market for the Common Shares; |
| ● | limited amount of news and analyst coverage; and |
| ● | a decreased ability to issue additional securities or obtain
additional financing in the future. |
Failure to comply with Nasdaq
rules, the delisting of NLS’s securities from Nasdaq or the inability to complete the Merger could materially adversely affect the
business, financial results and share price of NLS.
Following the closing of the Merger, NLS
may lose its foreign private issuer status, which would then require it to comply with the domestic reporting requirements of the Exchange Act,
and cause it to incur significant legal, accounting and other expenses.
In order to maintain NLS’s
status after the completion of the Merger as a foreign private issuer, either (i) a majority of its common shares must be either
directly or indirectly owned of record by non-residents of the United States; or (ii) (a) a majority of its executive officers
or directors may not be United States citizens or residents, (b) more than 50% of its assets cannot be located in the United States
and (c) its business must be administered principally outside the United States. If it lost this status, it would be required
to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed
and extensive than the requirements for foreign private issuers. Among other things, NLS would be required under current SEC rules to
prepare its financial statements in accordance with generally accepted accounting principles in the United States, which would involve
significant time and cost and could result in variations, which could be material, between historical financial results and as reported
under U.S. GAAP. It may also be required to make changes in its corporate governance practices in accordance with various SEC
and Nasdaq rules. The regulatory and compliance costs to NLS under U.S. securities laws if it is required to comply with the reporting
requirements applicable to a U.S. domestic issuer may be significantly higher than the cost it would incur as a foreign private issuer.
As a result, NLS expects that a loss of foreign private issuer status would increase NLS legal and financial compliance costs and would
make some activities highly time-consuming and costly. If it loses its foreign private issuer status and is unable to devote adequate
funding and the resources needed to maintain compliance with U.S. securities laws, while continuing its operations, NLS could be
forced to deregister with the SEC. A deregistration would substantially reduce or effectively terminate the trading of its securities
in the United States. We also expect that if NLS were required to comply with the rules and regulations applicable to U.S. domestic
issuers, it would make it more difficult and expensive for it to obtain director and officer liability insurance, and it may be required
to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more
difficult for NLS to attract and retain qualified directors.
Failure to consummate the Merger as currently
contemplated or at all could adversely affect the price of common shares and the future business and financial results of NLS.
The Merger may be consummated
on terms different than those contemplated by the Merger Agreement, or the Merger may not be consummated at all. If the Merger is not
completed, or is completed on different terms from those contemplated by the Merger Agreement, NLS could be adversely affected and subject
to a variety of risks associated with the failure to consummate the Merger, or to consummate the Merger as contemplated by the Merger
Agreement, including the following:
| ● | NLS’s shareholders may be prevented from realizing the
anticipated benefits of the Merger; |
| ● | the market price of common shares could decline significantly; |
| ● | reputational harm due to the adverse perception of any failure
to successfully consummate the Merger; |
| ● | incurrence of substantial costs relating to the proposed Merger,
such as legal, accounting, financial advisor, filing, printing and mailing fees, and, in certain cases, the payment by NLS of a termination
fee to Kadimastem; and |
| ● | the attention of NLS’s management and employees may
be diverted from their day-to-day business and operational matters as a result of efforts relating to attempting to consummate the
Merger. |
Any delay in the consummation
of the Merger or any uncertainty about the consummation of the Merger on terms other than those contemplated by the Merger Agreement,
or if the Merger is not completed, could materially adversely affect the business, financial results and share price of NLS.
The ownership interests of NLS’s will
be diluted by the consummation of the Merger, and NLS’s shareholders will exercise less influence over management than they exercised
before the Merger.
NLS’s shareholders have
the right to vote in the election of the NLS Board members and on certain other matters affecting NLS, as specified in NLS Bylaws, and
Kadimastem’s shareholders have the right to vote in the election of the Kadimastem Board and on certain other matters affecting
Kadimastem, as specified in Kadimastem’s articles of association. As a result of the Merger, the ownership position of existing
shareholders of NLS will decrease and Kadimastem’s shareholders will have an ownership position in NLS that is smaller than their
current stake in Kadimastem. The parties currently estimate the fully diluted share split at the Closing will be 80% to Kadimastem shareholders
and 20% to NLS shareholders. Consequently, NLS’s and Kadimastem shareholders will have less influence over the management and policies
of NLS after the Effective Time (as such term is defined below in “Item 4. Information on the Company — A. History and
Development of the Company — Agreement and Plan of Merger with Kadimastem”) than they currently exercise over the management
and policies of NLS and Kadimastem, respectively.
NLS’s and Kadimastem’s
directors and executive officers may have interests in the Merger that are different from, or in addition to, the interests of NLS’s
and Kadimastem’s shareholders generally. Mr. Alexander Zwyer will continue to be a director of the combined company after the
Effective Time of the Merger for a period of 12 months. Mr. Eric Konofal will continue to be a part-time officer of the
combined company after the Effective Time of the Merger. In addition, as of the Closing, NLS shall, at Kadimastem’s expense (up
to a maximum of $200,000), obtain a “run-off” prepaid directors’ and officers’ liability insurance policy for
the benefit of NLS’s current and former officers and directors, effective as of the Closing, with a reporting period of six (6)
years after the Closing, covering events, acts and omissions occurring before the Closing Date, and with coverage and amounts, and terms
and conditions that are acceptable to NLS. The premium for such policy (up to a maximum of $200,000) shall be paid by Kadimastem on or
prior to the Closing, and NLS shall take all necessary actions, and not fail to take any action, to prevent the cancellation of such policy
during its term.
Additionally, following the
Merger, several agreements related to compensation for NLS’ officers and directors will go into effecting, including:
| ● | Mr. Twito’s annual salary of USD 370,188
(including social benefits) which will increase to USD 413,743 upon the combined company completing a capital raise greater than $10 million
following the Merger and a bonus payment, subject to the discretion of the Board; and |
| ● | Each director on the board of NLS (1) shall receive
an annual amount of $25,000, payable quarterly, and a one-time option grant equal to 0.2% of the outstanding common shares, with
the initial vesting over three (3) years, with an exercised price equal to the market price at the Closing; and (2) will enter into indemnification
agreements between NLS and members of the board of directors. |
Additionally, Prof. Michel Revel, MD, Director
and Chief Scientific Officer of Kadimastem and the beneficial owner of 1,371,052 shares of Kadimastem, and Ronen Twito, Executive
Chairman and Chief Executive Officer, or CEO, of Kadimastem, and beneficial owner of 367,332 of Kadimastem, will benefit directly from
the exchange of their shares of Kadimastem in exchange for shares of NLS resulting from the Merger.
This may create potential conflicts of interest
or the appearance of such conflicts, which may lead to increased dissident shareholder activity, including litigation, which could result
in significant costs for NLS and Kadimastem and could materially delay or prevent the completion of the Merger.
The interests of NLS’s and Kadimastem’s
directors and executive officers include, among other things, the continued service as a director or executive officer of NLS following
the Merger and certain rights to continuing indemnification and directors’ and officers’ liability insurance for Kadimastem
directors and executive officers. There is a risk that these interests may influence the directors and executive officers to support the
Merger.
The interests of NLS’s
and Kadimastem’s directors and executive officers in the Merger may increase the risk of litigation intended to enjoin or prevent
the Merger and the risk of other related dissident shareholder activity. In the past, and in particular following the announcement of
a significant transaction, periods of volatility in the overall market or declines in the market price of a company’s securities,
shareholder litigation and dissident shareholder proposals have often been instituted against companies alleging conflicts of interest
in business dealings with affiliated or related persons and entities. For example, following the Merger, members of the combined company
Board will be entitled to new compensation terms, and certain officers of Kadimastem holding shares in Kadimastem will exchange such shares
for shares of NLS in connection with the Merger. The relationships described above may precipitate such activities by dissident shareholders
and, if instituted against NLS or Kadimastem or their respective directors or executive officers, such activities could result in substantial
costs, a material delay or prevention of the Merger and a diversion of management’s attention, even if the shareholder action is
without merit or unsuccessful.
The Merger is subject to the satisfaction
or waiver of conditions that may not be satisfied or completed on a timely basis, if at all. Failure to consummate the Merger could adversely
affect the future business and financial results of NLS.
The consummation of the Merger
is subject to the satisfaction or waiver of a number of conditions, including, among others, that the representations and warranties of
the parties are true and correct in all respects as of the closing date, NLS has at least $600,000 in gross funds (including cash in any
of its bank accounts) plus any proceeds received by NLS from certain parties in connection with its financing transactions undertake after
the execution of the Merger Agreement, Kadimastem has at least $3,500,000 in gross funds (including cash in any of its bank accounts)
minus any proceeds received by NLS from certain parties in connection with its financing transactions undertaken after the execution of
the Merger Agreement, Kadimastem’s receipt of certain tax rulings provided by Israeli Tax Authority and ISA, and approval of the
Merger Agreement and terms of the Merger by Kadimastem’s shareholders. These conditions make the completion, and the timing of the
completion, of the Merger uncertain. Kadimastem has already received a draft tax ruling from the Israeli Tax Authority and believes that
it will obtain the final ruling prior to NLS’s shareholders’ approval of the Merger. Further, the shareholders of Kadimastem
have approved the Merger.
However, NLS and Kadimastem
cannot provide assurance that the Merger will be consummated on the terms or timeline currently contemplated, or at all. If the Merger
is not completed on a timely basis, or at all, NLS may be adversely affected and subject to a number of risks, including the time and
resources committed by NLS’s respective management to matters relating to the Merger could otherwise have been devoted to pursuing
other opportunities.
NLS or Kadimastem may waive one or more
of the conditions to the Merger without re-soliciting shareholder approval.
NLS or Kadimastem may determine
to waive, in whole or in part, one or more of the conditions to its obligation to consummate the Merger. Any determination whether to
waive any condition to the Merger and whether to re-solicit shareholder approval or amend the related proxy statement/prospectus or Registration
Statement on Form F-4 as a result of a waiver will be made by NLS or Kadimastem, as applicable, at the time of such waiver based on the
facts and circumstances as they exist at that time.
The Merger may be completed even though
material adverse changes subsequent to the announcement of the Merger, such as industry-wide changes or other events, may occur.
In general, the parties
to the Merger can refuse to complete the Merger if there is a material adverse change affecting the other party. However, some types
of changes do not permit NLS and Kadimastem to refuse to complete the Merger, even if such changes would have a material adverse
effect on any of the parties involved in the Merger. For example, if there are changes in general national or international
economic, financial, political or business conditions, NLS and Kadimastem would not have the right to refuse to complete the Merger.
If adverse changes occur that affect the Merger but the parties are still required to complete the Merger, NLS’s share price,
business and financial results after the completion of the Merger may suffer.
NLS will have limited protection in the
event that any of the representations and warranties made by Kadimastem ultimately proves to be inaccurate or incorrect.
NLS will have limited protection
if any representation or warranty made by Kadimastem in the Merger Agreement proves to be inaccurate or incorrect, and such representations
and warranties will not survive the closing. Accordingly, to the extent such representations or warranties are incorrect, NLS would have
limited or no indemnification claims with respect thereto, may not recover any damages it may have suffered, and may not have sufficient
cash on hand or other resources to seek to pursue an alternative strategic transaction or avoid the dissolution and liquidation of NLS
in the event that the Merger does not close.
Each of NLS and Kadimastem prior to the
closing of the Merger expects to, and NLS following the closing of the Merger may, incur significant costs in connection with the Merger.
Each of NLS and Kadimastem
prior to the closing of the Merger expects to, and NLS following the closing of the Merger may, incur significant costs in connection
with the Merger, and may incur other unanticipated costs. While each company has assumed that a certain level of transaction and integration
expenses will be incurred, there are factors beyond each company’s control that could affect the total amount or the timing of those
expenses. Many of the expenses that may be incurred, by their nature, are difficult to estimate accurately at the current time. Each of
NLS and Kadimastem expect that the elimination of duplicative costs, as well as the realization of other efficiencies related to the integration
of the two businesses, may allow NLS to offset those incremental expenses over time, the net benefit of which may not be achieved in the
near term, or at all.
Lawsuits may be commenced seeking to enjoin
or prevent the Merger or seeking other relief which may delay or prevent the completion of the Merger and result in NLS or Kadimastem
incurring substantial costs.
Public company merger and
acquisition transactions are often subject to lawsuits initiated by plaintiffs seeking to enjoin or prevent the transaction or obtain
other relief. NLS, Kadimastem and their respective executive officers, directors and advisors may become subject to similar litigation
with respect to the Merger. Any such lawsuit could seek, among other things, injunctive or other equitable relief, including a request
to rescind parts of the Merger Agreement and otherwise to enjoin the parties from consummating the Merger, as well as to require payment
of fees and other costs by the defendants. NLS and Kadimastem may incur substantial costs defending any such lawsuit, including the distraction
of management’s attention, even if such lawsuits are without merit or unsuccessful. No assurance can be made as to the outcome of
any such lawsuits. If plaintiffs are successful in obtaining an injunction prohibiting the parties from completing the Merger or in obtaining
other relief, the completion of the Merger may be prevented or delayed or its terms could change.
If the Merger’s benefits do not meet
the expectations of investors, shareholders or financial analysts, the market price of securities may decline.
If the benefits of the Merger
do not meet the expectations of investors or securities analysts, the market price of common shares prior to the completion of the Merger
may decline. The market values of our securities at the time of the Merger may vary significantly from their prices on the date the Merger
Agreement was executed.
In addition, following the
Merger, fluctuations in the price of common shares could contribute to the loss of all or part of your investment. If an active market
for common shares develops and continues, the trading price of common shares following the Merger could be volatile and subject to wide
fluctuations in response to various factors, some of which are beyond our control. If the benefits of the Merger do not meet the expectations
of investors or securities analysts it could have a material adverse effect on your investment in common shares and common shares may
trade at prices significantly below the price you paid for them. In such circumstances, the trading price of common shares may not recover
and may experience a further decline.
Even if the Merger is completed, there is
no guarantee that any proceeds will be paid to NLS shareholders under the CVR Agreement.
Even if the Merger is completed,
there can be no assurance that any payments will be made to NLS shareholders under the CVR Agreement. The Contingent Value Rights provide
for potential payments to NLS shareholders based on the achievement of certain post-merger performance or milestone criteria. These criteria
depend on factors that are largely outside our control, such as the successful integration of the companies, the realization of anticipated
synergies, and the achievement of specific operational or financial targets within specified timeframes.
In addition, various risks,
uncertainties, and assumptions could impact the ability of the combined company to generate the necessary funds for such payments. These
include, but are not limited to, changes in market conditions, competitive pressures, regulatory developments, or unexpected costs that
may arise following the Merger. If the combined company is unable to achieve the necessary milestones or if additional unforeseen challenges
arise, it is possible that no payments will be made to NLS shareholders under the CVR Agreement.
Consequently, NLS shareholders
should understand that the CVR Agreement does not guarantee any future payments, and they may receive little or no proceeds from the CVR
Agreement in connection with the Merger.
Risks Related to NLS After the Consummation
of the Merger
Following the Merger, NLS intends to shift
its business focus to developing and manufacturing “off-the-shelf”, allogenic, proprietary cell products, which may not be
successful.
Following the Merger, NLS
intends to shift its business focus to developing and manufacturing “off-the-shelf”, allogenic, proprietary cell products,
which may not be successful. Planned operations and to grow and compete will depend on the availability of adequate capital. NLS cannot
assure you that it will be able to obtain equity or debt financing on acceptable terms, or at all, to adopt its new business, its planned
operations and to implement its growth strategy. As a result, NLS cannot assure you that adequate capital will be available to continue
its normal and planned operations and to finance its current growth plans, take advantage of business opportunities, or respond to competitive
pressures, any of which could harm its business.
Following the Merger, NLS may be unable
to integrate successfully and realize the anticipated benefits of the Merger.
The Merger involves the combination
of two companies which currently operate as independent companies. NLS may fail to realize some or all of the anticipated benefits of
the Merger if the integration process takes longer than expected or is more costly than expected.
Potential difficulties NLS
may encounter in the integration process include the following:
| ● | the inability to successfully combine the businesses of NLS
and Kadimastem in a manner that permits NLS to achieve the anticipated benefits from the Merger, which would result in the anticipated
benefits of the Merger not being realized partly or wholly in the time frame currently anticipated or at all; |
| ● | creation of uniform standards, controls, procedures, policies
and information systems; and |
| ● | potential unknown liabilities and unforeseen increased expenses,
delays or regulatory conditions associated with the Merger. |
In addition, NLS and Kadimastem
have operated and, until the completion of the Merger, will continue to operate, independently. It is possible that the integration process
also could result in the diversion of each company’s management’s attention, the disruption or interruption of, or the loss
of momentum in, each company’s ongoing businesses or inconsistencies in standards, controls, procedures and policies, any of which
could adversely affect NLS’s ability to maintain its business relationships or the ability to achieve the anticipated benefits of
the Merger, or could otherwise adversely affect the business and financial results of NLS.
Following the Merger, NLS’s business
strategy will depend heavily on advancing and commercializing its pipeline products. However, NLS’s research and development efforts
are subject to substantial risk, as drug development requires significant investment and faces inherent uncertainties.
Clinical trials may fail or
be delayed, regulatory approvals are uncertain, and even if NLS does obtain required regulatory approvals, commercial success is not guaranteed.
These risks could increase NLS’s post-Merger costs, delay potential revenues, and impact NLS’s ability to meet financial targets.
Any setbacks in research and development could materially reduce the anticipated benefits of the Merger and impact NLS’s financial
position and future growth potential. Furthermore, intensified research and development efforts may divert resources from other strategic
initiatives, limiting NLS’s ability to respond to unforeseen challenges and competitive pressures in a timely manner.
The future financial results of NLS will
suffer if NLS does not effectively manage its assets or deploy its available capital following the Merger.
Following the Merger, NLS
intends to focus on developing and manufacturing “off-the-shelf”, allogeneic, proprietary cell products. Future financial
results of NLS will suffer if NLS does not effectively manage this shift in business focus. If NLS is unable to obtain capital necessary
to maintain and increase its assets, and the assets of NLS, NLS could be required to reduce or suspend its operations or dispose of assets
at inopportune time or price, which could negatively affect NLS’s financial condition, results of operations and ability to pay
or sustain dividends to NLS’s shareholders.
NLS and Kadimastem have each had a history
of net losses, and NLS expects to continue to incur losses for the foreseeable future, including following the Merger. If NLS ever achieves
profitability, it may not be able to sustain it.
Each of NLS and
Kadimastem have incurred losses since its respective inception and expects to continue to incur losses for the foreseeable future,
including, with respect to NLS, following the Merger. NLS’s net loss attributable to holders of our common shares for the
years ended December 31, 2024 and 2023, respectively was approximately $4.1 million and $12.2 million. As of
December 31, 2024 and December 31, 2023 NLS had an accumulated deficit of approximately $74.4 million and
$70.4 million, respectively, and total equity of $1.4 million and $(8.8) million. Substantially all of our operating losses
resulted from costs incurred in connection with our clinical development program and from general and administrative costs
associated with our operations. Kadimastem reported net losses of approximately $7.2 million, $3.3 million and
$6.8 million for the years ended December 31,2024, 2023 and 2022, respectively, As of December 31, 2024 and
2023, Kadimastem had a total equity of approximately $(8.0) million and $(1.2) million, respectively, and accumulated deficit of
approximately $(76.5) million and $(69.3) million, respectively. NLS expects to incur net losses from continuing operations and
net cash used in operating activities, including following the Merger. NLS may need to raise additional working capital to continue
its normal and planned operations. NLS will need to generate and sustain significant revenue levels in future periods in order to
become profitable, and, even if NLS does, NLS may not be able to maintain or increase its level of profitability. NLS anticipates
that its operating expenses will remain substantially consistent in the foreseeable future. This reflects a decrease in operating
costs associated with discontinued assets as part of the anticipated merger, offset by increased consultancy efforts, acquisition
activities, and expanded marketing and sales initiatives aimed at growing its customer and client base. These expenditures will make
it necessary for NLS to continue to raise additional working capital and make it harder for us to achieve and maintain
profitability. NLS’s efforts to grow NLS’s business may be costlier than NLS expects, and NLS may not be able to
generate sufficient revenue to offset NLS’s increased operating expenses.
If NLS is forced to reduce
NLS’s operating expenses, NLS’s growth strategy could be compromised. NLS may incur significant losses in the future for a
number of reasons, including unforeseen expenses, difficulties, complications and delays and other unknown events. Accordingly, substantial
doubt exists about NLS’s ability to continue as a going concern, including following the Merger, and NLS cannot assure you that
NLS will achieve sustainable operating profits as NLS continues to expand NLS’s infrastructure, further develop NLS’s marketing
efforts, and otherwise implement NLS’s growth initiatives. The net losses that NLS incurs may fluctuate significantly from period
to period. NLS will need to generate significant additional revenue to achieve and sustain profitability. Even if NLS achieves profitability,
it cannot be sure that it will remain profitable for any substantial period of time.
NLS’s ability to grow and compete
in the future will be adversely affected if adequate capital is not available to it or not available on terms favorable to it.
The ability of NLS to continue
its normal and planned operations and to grow and compete will depend on the availability of adequate capital. NLS cannot assure you that
it will be able to obtain equity or debt financing on acceptable terms, or at all, to continue its normal and planned operations and to
implement its growth strategy. As a result, NLS cannot assure you that adequate capital will be available to continue its normal and planned
operations and to finance its current growth plans, take advantage of business opportunities, or respond to competitive pressures, any
of which could harm its business.
NLS will need substantial additional funding
to continue its operations, which could result in dilution to its shareholders. NLS may not be able to raise capital when needed, if at
all, which could cause it to have insufficient funds to pursue its operations, or to delay, reduce or eliminate its development of new
programs or commercialization efforts.
NLS expects to incur additional
costs associated with continuing to operate as a public company and to require substantial additional funding to continue to pursue its
business and continue with its expansion plans. NLS may also encounter unforeseen expenses, difficulties, complications, delays and other
unknown factors that may increase its capital needs and/or cause it to spend its cash resources faster than expected. Accordingly, NLS
expects that it will need to obtain substantial additional funding in order to continue its operations. To date, NLS has financed its
operations entirely through equity and debt investments by founders and other investors and the incurrence of debt, and it expects to
financing its operations through equity and debt investments by investors in the foreseeable future, including following the Merger. Additional
funding from those or other sources may not be available when or in the amounts needed, on acceptable terms, or at all. If NLS raises
capital through the sale and issuance of equity, or securities convertible into equity, it would result in dilution to its existing shareholders,
which could be significant depending on the price at which it may be able to sell and issue its securities. If it raises additional capital
through the incurrence of additional indebtedness, it would likely become subject to further covenants that could restricting its business
activities, and holders of debt instruments will likely have rights and privileges senior to those of its equity investors. In addition,
servicing the interest and principal repayment obligations under debt facilities could divert funds that would otherwise be available
to support development of new programs and marketing to current and potential new clients. If NLS is unable to raise capital when needed
or on acceptable terms, it could be forced to delay, reduce or eliminate development of new programs or future marketing efforts. Any
of these events could significantly harm NLS’s business, financial condition and prospects.
Without obtaining adequate capital funding
or improving its financial performance, NLS may not be able to continue as a going concern.
The report of NLS’s
independent registered public accounting firm on its consolidated financial statements as of and for the year ended December 31,
2024 includes an explanatory paragraph indicating that there is substantial doubt about its ability to continue as a going concern. If
it is unable to raise sufficient capital when needed, its business, financial condition and results of operations will be materially and
adversely affected, and it will need to significantly modify its operational plans to continue as a going concern. The inclusion of a
going concern explanatory paragraph by NLS’s auditors, its lack of cash resources and its potential inability to continue as a going
concern may materially adversely affect its share price and its ability to raise new capital or to enter into critical contractual relations
with third parties. If it is unable to continue as a going concern, including following the Merger, it might have to liquidate its assets
and the values it receives for its assets in liquidation or dissolution could be significantly lower than the values reflected in its
financial statement.
Increased operating and capital costs could
affect NLS’s profitability.
Costs for any particular
product are subject to variation due to a number of factors, such as regulatory costs and research and development expenses. In
addition, costs are affected by the price and availability of input commodities, electricity, labor, chemical reagents, and
processing related equipment and facilities. Product costs are, at times, subject to volatile price movements, including increases
that could make production at certain operations less profitable. Further, changes in laws and regulations can affect product
prices, uses and transport. Reported costs may also be affected by changes in accounting standards. A material increase in costs
could have a significant effect on NLS’s profitability and operating cash flow.
NLS could have significant
increases in capital and operating costs over the next several years in connection with the development of new projects and in the
sustaining and/or expansion of existing operations. Costs associated with capital expenditures may increase in the future as a result
of factors beyond NLS’s control. Increased capital expenditures may have an adverse effect on the profitability of and cash flow
generated from existing operations, as well as the economic returns anticipated from new projects.
NLS may seek to grow through acquisitions.
NLS may seek to grow through
acquisitions. Factors which may affect its ability to grow successfully through acquisitions include:
| ● | inability to obtain financing; |
| ● | difficulties and expenses in connection with integrating the
acquired companies and achieving the expected benefits; |
| ● | diversion of management’s attention from current operations; |
| ● | the possibility that it may be adversely affected by risk
factors facing the acquired companies; |
| ● | acquisitions could be dilutive to earnings, or in the event
of acquisitions made through the issuance of common shares to the shareholders of the acquired company, dilutive to the percentage of
ownership of its existing shareholders; |
| ● | potential losses resulting from undiscovered liabilities of
acquired companies not covered by the indemnification that it may obtain from the seller; and |
| ● | loss of key employees of the acquired companies. |
The potential termination of the Merger
Agreement and related uncertainties could negatively impact NLS’s business.
The Merger Agreement is subject
to several conditions, including approval by NLS’s shareholders, which must be met or waived prior to the Merger’s completion.
Some conditions are outside of NLS’s control, and there is no assurance that they will be met in a timely manner or at all. The
Merger Agreement could be delayed, may not be completed, or may be terminated under certain circumstances. Uncertainty regarding the Merger
could adversely impact employee morale and retention, disrupt business relationships, and affect the trading price of common shares. Additionally,
management and personnel will need to focus significant time and resources on the Merger, potentially diverting attention from day-to-day
operations. NLS will incur significant transaction costs regardless of whether the Merger is finalized, and such expenses may be higher
than anticipated, especially if the Merger is delayed or terminated.
Unfavorable general economic conditions
may materially adversely affect NLS’s business.
While it is difficult
for NLS to predict the impact of general economic conditions on its business, these conditions could reduce customer demand for some
of its products or services which could cause its revenue to decline. Also, NLS’s customers that are especially reliant on the
credit and capital markets may not be able to obtain adequate access to credit or equity funding, which could affect their ability
to make timely payments to NLS. Moreover, NLS relies on obtaining additional capital and/or additional funding to provide
working capital to support its operations. NLS regularly evaluates alternative financing sources. Further changes in the commercial
capital markets or in the financial stability of its investors and creditors may impact the ability of its investors and creditors
to provide additional financing. For these reasons, among others, if the economic conditions stagnate or decline, NLS’s
operating results and financial condition could be adversely affected.
Economic and political conditions may cause
fluctuations of the future prices, supply, and demand for products NLS and Kadimastem use, which may negatively affect NLS’s revenues.
Revenues generated from NLS’s
production activities in the biotech industry will be highly dependent upon the future prices and demand for products that NLS and Kadimastem
currently use. Factors which may affect prices and demand for products that NLS and Kadimastem currently use include, but are not limited
to, the worldwide supply of these products; the price of these products that are produced in the United States, Israel, Switzerland,
or imported from foreign countries; consumer demand for such products; the price and availability of alternative products; federal and
state regulation; and general, national and worldwide economic political conditions.
Changes in the regulatory environment could
have a material adverse effect on NLS’s business.
Changes in the regulatory
environment could have a material adverse effect on NLS’s business. NLS’s exploration, development, production and marketing
operations are subject to extensive environmental regulation at the federal, state and local levels including those governing pharmaceutical
research and testing. Under these laws and regulations, NLS could be held liable for personal injuries, medical malpractice and other
damages. Failure to comply with these laws and regulations may also result in the suspension or termination of NLS’s operations
and subject us to administrative, civil, and criminal penalties. Governmental laws and regulations also increase the costs to plan, permit,
design and install our operations and facilities.
Risks Related to Kadimastem’s Financial
Position and Capital Requirements
Kadimastem has incurred significant operating
losses since Kadimastem’s inception and anticipates that the combined company will incur continued losses for the foreseeable future.
Kadimastem is an
emerging biopharmaceutical company with a limited operating history. Kadimastem has funded its operations to date primarily through
raising capital on the Tel Aviv Stock Exchange (“TASE”), proceeds from the private placement of common shares, credit
facilities, loans and convertible notes. Kadimastem expects to continue to incur substantial losses over the next several years
during Kadimastem’s clinical development phase. To fully execute Kadimastem’s business plan, it will need to complete
Phase 3 clinical studies and certain development activities, as well as manufacture the required clinical and commercial
production batches in the pilot manufacturing plant. Further, Kadimastem’s product candidates will require regulatory approval
prior to commercialization, and it will need to establish sales, marketing and logistic infrastructures. These activities may span
many years and require substantial expenditures to complete and may ultimately be unsuccessful. Any delays in completing these
activities could adversely impact us. Management of Kadimastem plans to seek additional equity financing through private and public
offerings or strategic partnerships and, in the longer term, by generating revenues from product sales. Kadimastem has incurred
losses in each year since its inception. Kadimastem’s net loss attributable to holders of ordinary shares of Kadimastem, no
par value, or Kadimastem Ordinary Shares, for the years ended December 31, 2024 and 2023 were $7.2 million and
$3.3 million, respectively. Substantially all of Kadimastem’s operating losses resulted from costs incurred in connection
with Kadimastem’s development program and from general and administrative costs associated with Kadimastem’s
operations.
Until Kadimastem can generate significant
revenues, if ever, it expects to satisfy Kadimastem’s future cash needs through debt or equity financing. It cannot be certain that
additional funding will be available to it on acceptable terms, if at all. If funds are not available, Kadimastem may be required to delay,
reduce the scope of, or eliminate research or development plans for, or commercialization efforts with respect to Kadimastem’s products.
Kadimastem expects its research
and development expenses to increase in connection with its planned expanded clinical trials. In addition, if Kadimastem obtains marketing
approval for AstroRx® and/or IsletRx or any other current or future product candidate, it will likely incur significant
sales, marketing and outsourced manufacturing expenses, as well as continued research and development expenses. Furthermore, in the period
following this annual report, it expects to incur additional costs associated with operating as a public company, which Kadimastem estimates
will be at least several hundred thousand dollars annually. As a result, Kadimastem expects to continue to incur significant and increasing
operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing pharmaceutical
products, Kadimastem is unable to predict the extent of any future losses or when it will become profitable, if at all.
Furthermore, in addition to
such operating expenses, Kadimastem expects to incur additional costs associated with operating as a public company subject to the rules
and regulations of the SEC, which it estimates will be at least one million dollars annually. As a result, Kadimastem expects to continue
to incur significant and increasing operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated
with developing drug substances and product candidates, Kadimastem is unable to predict the extent of any future losses or when Kadimastem
will become profitable, if at all.
Kadimastem expects to continue
to incur significant losses until Kadimastem is able to commercialize Kadimastem’s product candidates, which it may not be successful
in achieving. Kadimastem anticipate that Kadimastem’s expenses will increase substantially if and as we:
| ● | continue the research and development of Kadimastem’s
product candidates; |
| ● | expand the scope of Kadimastem’s current clinical studies
for its product candidates; |
| ● | seek regulatory and marketing approvals for Kadimastem’s
product candidates that successfully complete clinical studies; |
| ● | establish a sales, marketing, and distribution infrastructure
to commercialize Kadimastem’s product candidates; |
| ● | seek to identify, assess, acquire, license, and/or develop
other product candidates and subsequent generations of Kadimastem’s current product candidates; |
| ● | seek to maintain, protect, and expand Kadimastem’s intellectual
property portfolio; |
| ● | seek to attract and retain skilled personnel; and |
| ● | create additional infrastructure to support Kadimastem’s
operations as a public company and Kadimastem’s product candidate development and planned future commercialization efforts. |
Kadimastem has not generated revenue from
any product candidate and may never be profitable, even if Kadimastem receives regulatory approval to commercialize its products in additional
geographical territories and indications.
Kadimastem’s ability
to become profitable depends upon Kadimastem’s ability to generate revenue. To date, Kadimastem have not generated any revenue from
Kadimastem’s development stage product candidates, AstroRx® and/or IsletRx. In order to generate significant revenue,
it will need to obtain additional regulatory approvals in jurisdictions within which it already has certain regulatory approvals, and
also in jurisdictions in which it currently has no regulatory approvals to market Kadimastem’s products. Even if Kadimastem’s
current products or any future products are approved for marketing and sale, it anticipates incurring significant incremental costs associated
with commercializing such products.
Kadimastem’s ability
to become profitable depends upon Kadimastem’s ability to generate revenue. To date, Kadimastem has not generated any revenue from
Kadimastem’s development stage product candidates, AstroRx® and/or IsletRx, and do not know when, or if, it will
generate any such revenue. Kadimastem does not expect to generate significant revenue unless or until Kadimastem obtains marketing approval
of, and commercializes, AstroRx® and/or IsletRx. Kadimastem’s ability to generate future revenue from product candidate
sales depends heavily on its success in many areas, including but not limited to:
| ● | obtaining favorable results from and progress the pre-clinical
and clinical development of Kadimastem’s product candidates, namely AstroRx and/or IsletRx; |
| ● | developing and obtaining regulatory approval for registration
studies protocols for Kadimastem’s product candidates, namely AstroRx® and/or IsletRx; |
| ● | subject to successful completion of registration and clinical
trials of AstroRx® and/or IsletRx, applying for and obtaining marketing approval; |
| ● | establishing and maintaining supply and manufacturing relationships
with third parties that can provide adequate (in amount and quality) products, and at acceptable costs, to support market demand for
Kadimastem’s product candidates, if marketing approval is received; |
| ● | identifying, assessing, acquiring and/or developing new product
candidates; |
| ● | accurately identifying demand for Kadimastem’s product
candidates; |
| ● | obtaining market acceptance of Kadimastem’s product
candidates, if approved for marketing, as viable treatment options; |
| ● | negotiating favorable terms in any collaboration, licensing
or other arrangements into which Kadimastem may enter; |
| ● | establishing and nurturing relationships with the leading
physicians in the United States; and |
| ● | attracting, hiring and retaining qualified personnel. |
Kadimastem does not believe that its current
cash on hand will be sufficient to fund its projected operating requirements. This raises substantial doubt about its ability to continue
as a going concern.
Kadimastem does not believe
that its current cash on hand will be sufficient to fund its projected operating requirements. This raises substantial doubt about Kadimastem’s
ability to continue as a going concern. If Kadimastem cannot continue as a going concern, its investors may lose their entire investment
in its common shares. Until Kadimastem can generate significant revenues, if ever, it expects to satisfy its future cash needs through
debt or equity financing. Kadimastem cannot be certain that additional funding will be available to it on acceptable terms, if at all.
If funds are not available, Kadimastem may be required to delay, reduce the scope of, or eliminate research or development plans for,
or commercialization efforts with respect to its products.
Even if the Merger is completed,
Kadimastem expects that it will need to raise substantial additional funding before it can expect to complete the development of AstroRx®
and IsletRx or any other product candidate. This additional financing may not be available on acceptable terms, or at all. Failure to
obtain this necessary capital when needed may force Kadimastem to delay, limit or terminate its product candidate development efforts
or other operations.
Even if the Merger is completed,
Kadimastem expects it will require substantial additional capital to commercialize its product candidates. In addition, its operating
plans may change as a result of many factors that may not currently be known to it, and it may need to seek additional funds sooner than
planned. Kadimastem’s future funding requirements will depend on many factors, including but not limited to:
| ● | its clinical trial results; |
| ● | the cost, timing and outcomes of seeking marketing approval
of AstroRx® and/or IsletRx; |
| ● | the cost of filing and prosecuting patent applications and
the cost of defending its patents; |
| ● | the cost of prosecuting patent infringement actions against
third parties; |
| ● | development of other early-stage development product candidates; |
| ● | the costs associated with commercializing AstroRx®
and/or IsletRx if Kadimastem receives marketing approval, including the cost and timing of establishing sales and marketing capabilities
to market and sell AstroRx® and/or IsletRx; |
| ● | subject to receipt of marketing approval, revenue received
from sales of approved products, if any, in the future; |
| ● | any product liability or other lawsuits related to Kadimastem’s
products; |
| ● | the expenses needed to attract and retain skilled personnel;
and |
| ● | the costs associated with being a public company. |
Any additional fundraising
efforts may divert its management from their day-to-day activities, which may adversely affect Kadimastem’s ability to develop
and commercialize its product candidates. In addition, Kadimastem cannot guarantee that future financing will be available in sufficient
amounts or on terms acceptable to it, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights
of holders of its securities and the issuance of additional securities, whether equity or debt, by it, or the possibility of such issuance,
may cause the market price of its common shares to decline. The incurrence of indebtedness could result in increased fixed payment obligations,
and Kadimastem may be required to agree to certain restrictive covenants, such as limitations on its ability to incur additional debt,
limitations on its ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely
impact its ability to conduct its business. Kadimastem could also be required to seek funds through arrangements with collaborative partners
or otherwise at an earlier stage than otherwise would be desirable, and it may be required to relinquish rights to some of its technologies
or product candidates or otherwise agree to terms unfavorable to it, any of which may have a material adverse effect on its business,
operating results and prospects. Even if it believes that it has sufficient funds for its current or future operating plans, Kadimastem
may seek additional capital if market conditions are favorable or if it has specific strategic considerations.
If Kadimastem is unable to
obtain funding on a timely basis, it may be required to significantly curtail, delay or discontinue one or more of its research or development
programs or the development or commercialization, if any, of any product candidates or be unable to expand its operations or otherwise
capitalize on its business opportunities, as desired, which could materially affect its business, financial condition and results of operations.
Unstable market and economic conditions,
including inflation, may have serious adverse consequences on Kadimastem’s business, financial condition and share price.
The global economy, including
credit and financial markets, has experienced extreme volatility and disruptions, including severely diminished liquidity and credit availability,
declines in consumer confidence, declines in economic growth, increases in unemployment rates, increases in inflation rates and uncertainty
about economic stability. The current wars in Israel and between Ukraine and Russia have created extreme volatility in the global capital
markets, and are expected to have further global economic consequences, including disruptions of the global supply chain and energy markets.
Any such volatility and disruptions may have adverse consequences on Kadimastem or the third parties on whom Kadimastem rely. If the equity
and credit markets deteriorate, including as a result of political unrest or war, it may make any necessary debt or equity financing more
difficult to obtain in a timely manner or on favorable terms, more costly or more dilutive.
Disruption to the global economy
could also result in a number of follow-on effects on Kadimastem’s business, including a possible slow-down resulting from lower
customer expenditures; inability of customers to pay for products, solutions or services on time, if at all; more restrictive export regulations,
which could limit Kadimastem’s potential customer base; negative impact on Kadimastem’s liquidity and financial condition
and share price, which may impact Kadimastem’s ability to raise capital in the market, obtain financing and secure other sources
of funding in the future on terms favorable to Kadimastem.
Inflation, which increased
significantly during 2024, could adversely affect Kadimastem’s business by increasing the costs of raw material and labor needed
to operate its business and could continue to adversely affect Kadimastem in future periods. If this current inflationary environment
continues, there can be no assurance that Kadimastem would be able to recover related cost increases through price increases, which could
result in downward pressure on Kadimastem’s operating margins. As a result, Kadimastem’s financial condition, results of operations,
and cash flows could be adversely affected over time.
Risks Related to Product Development, Regulatory
Approval and Commercialization
Kadimastem depends substantially on the
success of its proprietary product candidates. Kadimastem cannot give any assurance that any of its drug substances and product candidates
will receive regulatory approval, which is necessary before they can be commercialized.
Kadimastem has invested almost
all of its efforts and financial resources in research and development of Kadimastem’s drug substances, as defined by the Harmonised
Tripartite Guideline for Good Clinical Practice (ICH-GCP E6), and product candidates and general and administrative costs. Kadimastem’s
portfolio comprises a clinical program, AstroRx®, human astrocytes derived from pluripotent stem cells for the treatment
of neurodegenerative diseases such as amyotrophic lateral sclerosis (“ALS”), as well as a preclinical proof of concept program, IsletRx, human pancreatic islet like clusters
for the treatment of insulin dependent diabetes. The process to develop, obtain regulatory approval for and commercialize pharmaceutical
drug substances and product candidates is long, complex, costly, and inherently uncertain of outcome. Kadimastem is not permitted to
market any of its drug substances and product candidates in the United States, the EU, or any other jurisdiction until Kadimastem
receives the requisite regulatory approvals. Kadimastem cannot give any assurance that its current clinical development plan will proceed
as planned, that its product candidates will receive regulatory approval, or that such regulatory approval, if received, will be within
a timeframe that allows Kadimastem to effectively compete with its competitors or be successfully marketed and commercialized, which could
harm its business, operating results, prospects or financial condition.
All of Kadimastem’s
drug substances and product candidates are in various stages of clinical and/or preclinical development. Clinical drug development is
a lengthy and expensive process with uncertain timelines and uncertain outcomes. If clinical trials of its drug substances and product
candidates are prolonged, delayed or not commercially viable, Kadimastem or its collaborators may be unable to obtain required regulatory
approvals, and therefore may be unable to commercialize its drug substances and product candidates on a timely basis or at all, which
will adversely affect its business.
To obtain the requisite regulatory
approvals to market and sell any of Kadimastem’s product candidates, Kadimastem or its collaborators for such candidates must demonstrate
through extensive preclinical studies and clinical trials that its products are safe, pure and potent or effective in humans. Further,
the process of obtaining regulatory approval is expensive, often takes many years following the commencement of clinical trials and
can vary substantially based upon the type, complexity and novelty of the drug substances and product candidates involved, as well as
the target indications and patient population. Prior to obtaining approval to commercialize a product candidate in the United States
or abroad, Kadimastem or its potential future collaborators must demonstrate with substantial evidence from adequate and well-controlled
clinical trials, and to the satisfaction of the FDA, or comparable foreign regulatory authorities, that such drug substances and product
candidates are safe and effective for their intended uses. Additionally, clinical testing is expensive and can take many years to
complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process and its future clinical
trial results may not be successful.
Kadimastem may not be able
to commence or complete the clinical trials that would support its submission of a BLA to the FDA or an MAA to the EMA, and other regulatory
approvals from the Israeli Mistry of Health, or MOH. Drug development is a long, expensive and uncertain process, and delay or failure
can occur at any stage of any of its clinical trials. Clinical trials can be delayed or prevented for a number of reasons, including:
| ● | difficulties obtaining regulatory approval to commence a clinical
trial or complying with conditions imposed by a regulatory authority regarding the scope or term of a clinical trial; |
| ● | delays in reaching or failing to reach agreement on acceptable
terms with prospective contract research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation
and may vary significantly among different CROs and trial sites; |
| ● | insufficient or inadequate supply or quality of a product
candidate or other materials necessary to conduct its clinical trials; |
| ● | if the FDA or EMA elect to enact policy changes; |
| ● | difficulties obtaining institutional review board, or IRB,
approval to conduct a clinical trial at a prospective site; and |
| ● | challenges recruiting and enrolling patients to participate
in clinical trials for a variety of reasons, including size and nature of patient population, proximity of patients to clinical sites,
eligibility criteria for the trial, nature of trial protocol, the availability of approved effective treatments for the relevant disease
and competition from other clinical trial programs for similar indications. |
Clinical trials may also be
delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated
by Kadimastem, the FDA, the IRBs at the sites where the IRBs are overseeing a trial, a data safety monitoring board overseeing the clinical
trial at issue or by other regulatory authorities due to a number of factors, including:
| ● | failure to conduct the clinical trial in accordance with regulatory
requirements or its clinical protocols; |
| ● | inspection of the clinical trial operations or trial sites
by the FDA or other regulatory authorities; |
| ● | inspection of the drug substance manufacturing facility by
the FDA or other regulatory authorities; |
| ● | unforeseen safety issues or lack of effectiveness (futility);
and |
| ● | lack of adequate funding to continue the clinical trial. |
Any of these occurrences may
harm its business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in
the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of its drug substances
and product candidates or result in the development of its drug substances and product candidates being stopped early.
Kadimastem’s development
costs will increase if Kadimastem has material delays in its clinical trials, or if it is required to modify, suspend, terminate or repeat
a clinical trial. If Kadimastem is unable to conduct its clinical trials properly and on schedule, marketing approval may be delayed or
denied by the FDA, EMA, MOH, and other regulatory authorities.
Kadimastem cannot market and sell its cell
therapy drug substances and product candidates in the United States, Europe, or in other countries if it fails to obtain the necessary
regulatory approvals or licensure.
Kadimastem cannot sell its
cell therapy drug substances and product candidates until regulatory agencies grant marketing approval, or licensure. The process of obtaining
regulatory approval is lengthy, expensive, and uncertain. It is likely to take at least several years to obtain the required regulatory
approvals for its cell therapy product candidates, or it may never gain the necessary approvals.
Any difficulties that Kadimastem encounters
in obtaining regulatory approval may have a substantial adverse impact on its operations and cause its share price to decline significantly.
To obtain marketing approvals
in the United States and Europe for cell therapy drug substances and product candidates Kadimastem must, among other requirements,
complete carefully controlled and well-designed clinical trials sufficient to demonstrate to the FDA, the EMA and the PMDA that the cell
therapy drug substances and product candidates is safe and effective for each disease for which Kadimastem seeks approval. Several factors
could prevent completion or cause significant delay of its clinic trials, including an inability to enroll the required number of patients
or failure to demonstrate adequately that cell therapy drug substances and product candidates are safe and effective for use in humans.
Negative or inconclusive results from or adverse medical events during a clinical trial could cause the clinical trial to be repeated
or a program to be terminated, even if other studies or trials relating to the program are successful. The FDA or EMA can place a clinical
trial on hold if, among other reasons, it finds that patients enrolled in the trial are or would be exposed to an unreasonable and significant
risk of illness or injury. If safety concerns develop, Kadimastem, the FDA, the EMA or other regulatory bodies could stop its trials before
completion, which could harm its business, operating results, prospects or financial condition.
Obtaining approval of a BLA or a MAA even
after clinical trials that are believed to be successful is an uncertain process.
Kadimastem is not permitted
to market its products in the United States or the EU until it receives regulatory approval of a BLA from the FDA or MAA from the
EMA, or in any foreign countries until it receive the requisite approval from regulatory authorities in such countries.
Even if Kadimastem completes
its planned clinical trials and believe the results to be successful, all of which are uncertain, obtaining regulatory approval is an
extensive, lengthy, expensive and uncertain process, and the FDA and EMA, and other regulatory authorities may delay, limit or deny approval
of its products for many reasons, including, but not limited to:
| ● | Kadimastem may not be able to demonstrate to their satisfaction
that the product candidate is a safe or effective treatment for a given indication; |
| ● | the results of clinical trials may not meet the level of statistical
significance or clinical significance required by the regulatory agencies; |
| ● | disagreements regarding the number, design, size, conduct
or implementation of Kadimastem’s clinical trials, or with its interpretation of data from pre-clinical studies or clinical trials; |
| ● | a lack of acceptance of the accuracy or sufficiency of the
data generated at Kadimastem’s clinical trial sites to demonstrate, among others, that clinical and other benefits outweigh its
safety risks or to support the submission of a BLA or MAA; |
| ● | difficulties scheduling an advisory committee meeting in a
timely manner or the advisory committee, or such other similar committee, may recommend against approval of Kadimastem’s application
or may recommend that such regulators require, as a condition of approval, additional pre-clinical studies or clinical trials, improvements
in the manufacturing facility and stability transportation processes and durability limitations on approved labelling, or distribution
and use restrictions; |
| ● | the requirement that Kadimastem develop a REMS as a condition
of approval, which may or may not be feasible for Kadimastem; |
| ● | the identification of deficiencies in the manufacturing processes
in its manufacturing facility or facilities of third-party manufacturers with which Kadimastem enter into agreements for clinical and
commercial supplies; |
| ● | changes in approval policies or the adoption of new regulations
by such regulators; and |
| ● | Kadimastem may be unable to be granted a PIP deferral which
Kadimastem intends to request from the EMA for clinical trials in children; this may delay Kadimastem’s clinical trial program
or approvals for adults, or it may have successful clinical trial results for adults but not children (if Kadimastem were required to
conduct pediatric studies prior to the receipt of a BLA or MMA for use of its drug substances and product candidates in adults), or vice
versa. |
Before Kadimastem can submit
BLA to the FDA, Kadimastem must conduct Phase 3 clinical trials, that will be substantially broader than its Phase 2 trials.
A BLA must be supported by extensive clinical and pre-clinical data, as well as extensive information regarding chemistry, manufacturing
and controls to demonstrate the safety and effectiveness of the applicable product candidate. The number and types of pre-clinical studies
and clinical trials that will be required varies depending on the product candidate, the disease or condition that the product candidate
is designed to target and the regulations applicable to any particular product candidate. Obtaining approval of a BLA is a lengthy, expensive
and uncertain process, and Kadimastem may not be successful in obtaining approval. The FDA review processes can take years to complete
and approval is never guaranteed.
In this respect,
Kadimastem will also need to agree on a protocol with the FDA for the Phase 3 clinical trials before commencing those trials.
Phase 3 clinical trials frequently produce unsatisfactory results even though prior clinical trials were successful. Therefore,
the results of the additional trials that it conducts may or may not be successful. The FDA may suspend all clinical trials or
require that Kadimastem conducts additional clinical, nonclinical, manufacturing improvements, manufacturing validation or drug
substances quality studies and submit those data before it will consider or reconsider the BLA. Depending on the extent of
these or any other studies, approval of any applications that Kadimastem submit may be delayed by several years, or may require
it to expend more resources than it has available. It is also possible that additional studies, if performed and completed, may not
be considered sufficient by the FDA to approve the BLA. If any of these outcomes occur, Kadimastem would not receive approval
at such time, if any, that it seeks FDA approval. Kadimastem may face similar risks with respect to obtaining regulatory approval
from the EMA at such time, if any, that it seeks EMA approval. The risks that it faces in obtaining applicable approvals from the
FDA and EMA for AstroRx® and/or IsletRx, or any other product candidate that it may seek to develop, may also exist
with other regulatory authorities, such as those in Latin America or other regions.
Even if Kadimastem obtain
FDA, EMA or other regulatory approval for AstroRx® and/or IsletRx, the approval might contain significant limitations related
to use restrictions, warnings, precautions or contraindications, or may be subject to significant post-marketing studies or risk mitigation
requirements. If Kadimastem is unable to successfully commercialize AstroRx® and/or IsletRx, it may be forced to cease
operations.
Preliminary data that Kadimastem or others
announce or publish from time to time with respect to its products may change as more data become available and are subject to audit and
verification procedures that could result in material changes in the final data.
From time to time, Kadimastem,
or its partners, may publish or seek to publish preliminary data from ongoing clinical trials, which are based on a preliminary analysis
of then-available data. Positive preliminary data may not be predictive of such trial’s subsequent or overall results. Preliminary
data are subject to the risk that one or more of the results and related findings and conclusions may materially change following a more
comprehensive review of the data or as more data become available. Therefore, positive preliminary results in any ongoing clinical trial
may not be predictive of such results in the completed trial. Kadimastem also make assumptions, estimations, calculations and conclusions
as part of its analyses of data, and it may not have received or had the opportunity to fully evaluate all data. As a result, preliminary
data that it reports may differ from future results from the same clinical trials, or different conclusions or considerations may qualify
such results, once additional data have been received and fully evaluated. Preliminary data also remain subject to audit and verification
procedures that may result in the final data being materially different from the preliminary data it previously published. As a result,
preliminary data should be viewed with caution until the final data are available. Material adverse changes in the final data compared
to preliminary data could significantly harm its business prospects.
Further, others, including
regulatory agencies, may not accept or agree with its assumptions, estimates, calculations, conclusions or analyses or may interpret or
weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization
of the particular product candidate or product and its company in general. In addition, the information it chose to publicly disclose
regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with
what Kadimastem determines is material or otherwise appropriate information to include in its disclosure. If the interim, top-line or
preliminary data that Kadimastem reports differ from actual results, or if others, including regulatory authorities, disagree with the
conclusions reached, its ability to obtain approval for, and commercialize, in scale, its drug substances and product candidates may be
harmed, which could harm its business, operating results, prospects or financial condition.
The results of preclinical studies and early-stage
clinical trials of Kadimastem’s drug substances and product candidates may not be predictive of the results of later-stage clinical
trials. Initial success in Kadimastem’s ongoing clinical trials may not be indicative of results obtained when these trials are
completed or in later stage trials.
Drug substances and product
candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through
preclinical studies and initial clinical trials. Furthermore, there can be no assurance that any of Kadimastem’s clinical trials
will ultimately be successful or support further clinical development of any of Kadimastem’s product candidates. There is a high
failure rate for drugs and biologics proceeding through clinical trials. A number of companies in the pharmaceutical and biotechnology
industries have suffered significant setbacks in clinical development even after achieving promising results in earlier studies, and any
such setbacks in Kadimastem’s clinical development could harm its business and operating results.
The results of clinical trials conducted
at clinical sites outside the United States may not be accepted by the FDA and the results of clinical trials conducted at clinical
sites in the United States may not be accepted by international regulatory authorities.
Kadimastem plans to conduct
some of its clinical trials outside the United States. Such trials would be guided under FDA or EMA guidelines and inspections. It
is planning to globally develop AstroRx® and/or IsletRx in the United States and the EU. Although the FDA may
accept data from clinical trials conducted outside the United States, acceptance of this data is subject to certain conditions imposed
by the FDA. For example, the clinical trial must be well-designed and conducted and performed by qualified investigators in accordance
with ethical principles such as IRB or ethics committee approval and informed consent. The study population must also adequately represent
the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the
FDA deems clinically meaningful. Generally, the subject population for any clinical trials conducted outside of the United States
must be representative of the U.S. population. In addition, while these clinical trials are subject to the applicable local laws,
FDA acceptance of the data will be dependent upon its determination that the trials were conducted consistent with all applicable U.S. laws
and regulations. There can be no assurance the FDA or international regulatory authorities will accept data from trials conducted outside
of the United States or inside the United States, as the case may be, as adequate support of a marketing application. If the
FDA does not accept the data from sites in its globally conducted clinical trials, or if international regulatory authorities do not accept
the data from its U.S. clinical trials, it would likely result in the need for additional trials, which would be costly and time-consuming
and could delay or permanently halt the development of one or more of its product candidates.
The results of Kadimastem’s clinical
trials may not support its product candidates’ claims or any additional claims Kadimastem may seek for its drug substances and product
candidates and its clinical trials may result in the discovery of adverse side effects.
Even if any clinical trial
that Kadimastem needs to undertake is completed as planned, or if interim results from existing clinical trials are released, Kadimastem
cannot be certain that such results will support its drug substances and product candidates claims or any new indications that Kadimastem
may seek for its products or that the FDA or foreign authorities will agree with its conclusions regarding the results of those trials.
The clinical trial process may fail to demonstrate that its products or a product candidate is safe and effective for the proposed indicated
use, which could cause it to stop seeking additional clearances or approvals for its product candidates. Any delay or termination of Kadimastem’s
clinical trials will delay the filing of its regulatory submissions and, ultimately, Kadimastem’s ability to commercialize a product
candidate. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part
of the product candidate’s profile.
Kadimastem’s drug substances and product
candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the
commercial potential or result in significant negative consequences following regulatory approval, if obtained.
During the conduct of clinical
trials, patients may experience changes in their health, including illnesses, injuries, discomforts or a fatal outcome. It is possible
that as Kadimastem develops AstroRx® and/or IsletRx, or other drug substances and product candidates that Kadimastem may
seek to develop, in larger, longer and more extensive clinical trials as use of its product candidates becomes more widespread if they
receive regulatory approval, illnesses, injuries, discomforts and other adverse events that were observed in earlier clinical trials,
as well as conditions that did not occur or went undetected in previous clinical trials, will be reported by subjects. Many times, side
effects are only detectable after investigational products are tested in larger scale, Phase 2 and 3 clinical trials or, in some
cases, after they are made available to patients on a commercial scale after approval. If additional clinical experience indicates that
AstroRx® and/or IsletRx, or other drug substances and product candidates that Kadimastem may seek to develop, have side
effects or cause serious or life-threatening side effects, the development of the product candidate may fail or be delayed, or, if the
product candidate has received regulatory approval, such approval may be revoked or limited.
Additionally, if any of its
drug substances and product candidates receives marketing approval, the FDA or EMA could require it to adopt a REMS to ensure that the
benefits outweigh its risks, which may include, among other things, a medication guide outlining the risks of the product for distribution
to patients, a communication plan to health care practitioners, and restrictions on how or where the product can be distributed, dispensed
or used. Furthermore, if Kadimastem or others later identify undesirable side effects caused by AstroRx® and/or IsletRx,
several potentially significant negative consequences could result, including:
| ● | regulatory authorities may suspend or withdraw approvals of
such a product candidate; |
| ● | regulatory authorities may require additional warnings on
the label; |
| ● | regulatory authorities may issue negative publicity regarding
the affected product, including safety communications; |
| ● | Kadimastem may be required to change the way the product is
manufactured, distributed, dispensed or administered, or conduct additional pre-clinical studies or clinical trials; |
| ● | Kadimastem may need to voluntarily recall its products; and |
| ● | Kadimastem could be sued and held liable for harm caused to
patients. |
Any of these events could
prevent it from achieving or maintaining market acceptance of the affected product candidate and could significantly harm its business,
prospects, financial condition and results of operations.
Changes in methods of product candidate
manufacturing or formulation may result in additional costs or delay.
As drug substances and product
candidates proceed through pre-clinical studies to late-stage clinical trials towards potential approval and commercialization, it is
common that various aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an
effort to optimize processes and results and/or reduce cost of goods sold. Such changes carry the risk that they will not achieve these
intended objectives. Any of these changes could cause its drug substances and product candidates to perform differently and affect the
results of planned clinical trials or other future clinical trials conducted with the materials manufactured using altered processes.
Such changes may also require additional testing, FDA or EMA notification or FDA approval. This could delay completion of clinical trials,
require the conduct of bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay
approval of Kadimastem’s drug substances and product candidates and jeopardize its ability to commence sales and generate revenue.
Legislative or regulatory healthcare reforms
in the United States may make it more difficult and costly for the combined company to obtain regulatory clearance or approval of
its product candidates and to produce, market and distribute combined company’s products after clearance or approval
is obtained.
From time to time, legislation
is drafted and introduced in United States Congress, or Congress, that could significantly change the statutory provisions governing
the regulatory clearance or approval, manufacture and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations
and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect combined company’s business and
products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review
times of the combined company’s product candidates. NLS and Kadimastem cannot determine what effect changes in regulations, statutes,
legal interpretation or policies, when and if promulgated, enacted or adopted may have on the combined company’s business in the
future. Such changes could, among other things, require:
| ● | changes to manufacturing methods; |
| ● | change in protocol design; |
| ● | additional treatment arm (control); |
| ● | recall, replacement, or discontinuance of one or more of the
combined company’s products; and |
| ● | additional recordkeeping. |
In addition, in the
United States, there have been a number of legislative and regulatory proposals to change the health care system in ways that
could affect the combined company’s ability to sell the combined company’s products profitably. The pharmaceutical
industry in the United States, as an example, has been affected by the passage of the Patient Protection and Affordable Care
Act, or Affordable Care Act, which, among other things, imposed new fees on entities that manufacture or import certain branded
prescription drugs and expanded pharmaceutical manufacturer obligations to provide discounts and rebates to certain government
programs. There have been executive, judicial and Congressional challenges to certain aspects of the Affordable Care Act. For
example, on June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the Affordable
Care Act is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. On
August 16, 2022, President Biden signed the Inflation Reduction Act of 2022, or IRA, into law, which among other
things, extends enhanced subsidies for individuals purchasing health insurance coverage in Affordable Care Act marketplaces through
plan year 2025. The IRA also eliminates the “donut hole” under the Medicare Part D program beginning in 2025 by
significantly lowering the beneficiary maximum out-of-pocket cost and creating a new manufacturer discount program. It is possible
that the Affordable Care Act will be subject to judicial or Congressional challenges in the future. It is unclear how any such
challenges and any potential future healthcare reform measures of the Trump administration will impact the Affordable Care Act and
the combined company’s business.
Other legislative changes
have been proposed and adopted in the United States since the Affordable Care Act was enacted. These changes include, among others,
aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect in 2013 and, due to subsequent
legislative amendments to the statute, will remain in effect until 2032 unless additional Congressional action is taken. Congress is considering
additional health reform measures.
Further, there has been particular
and increasing legislative and enforcement interest in the United States with respect to drug pricing practices in recent years,
particularly with respect to drugs that have been subject to relatively large price increases over relatively short time periods. There
have been several recent U.S. Presidential executive orders, Congressional inquiries and proposed bills designed to, among other
things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between
pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs.
For example, in July 2021,
the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions
aimed at prescription drugs. In response to President Biden’s executive order, on September 9, 2021, the Department of Health
and Human Services, or HHS, released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform
and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can
take to advance these principles. Further, the IRA, among other things (i) directs HHS to negotiate the price of certain high-expenditure,
single-source drugs and biologics covered under Medicare and (ii) imposes rebates under Medicare Part B and Medicare Part D
to penalize price increases that outpace inflation. These provisions will take effect progressively starting in fiscal year 2023. On August 29,
2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the Medicare drug price negotiation
program is currently subject to legal challenges. On January 17, 2025, HHS selected fifteen additional drugs covered under Part D
for price negotiation in 2025. Each year thereafter more Part B and Part D products will become subject to the Medicare Drug
Price Negotiation Program. It is currently unclear how the IRA will be implemented but is likely to have a significant impact on the pharmaceutical
industry. Further in response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released
a report outlining three new models for testing by the Centers for Medicare & Medicare Services, or CMS, Innovation Center which
will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether
the models will be utilized in any health reform measures in the future.
Further, on December 7,
2023, the Biden administration announced an initiative to control the price of prescription drugs through the use of march-in rights under
the Bayh-Dole Act. On December 8, 2023, the National Institute of Standards and Technology published for comment a Draft Interagency
Guidance Framework for Considering the Exercise of March-In Rights which for the first time includes the price of a product as one factor
an agency can use when deciding to exercise march-in rights. While march-in rights have not previously been exercised, it is uncertain
if that will continue under the new framework. Individual states in the United States have also become increasingly active in passing
legislation and implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement
constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and in some cases,
designed to encourage importation from other countries and bulk purchasing. For example, on January 5, 2024, the FDA approved Florida’s
Section 804 Importation Program (SIP) proposal to import certain drugs from Canada for specific state healthcare programs. It is
unclear how this program will be implemented, including which drugs will be chosen, and whether it will be subject to legal challenges
in the United States or Canada. Other states have also submitted SIP proposals that are pending review by the FDA. Any such
approved importation plans, when implemented, may result in lower drug prices for products covered by those programs.
In the future, there will
likely continue to be proposals relating to the reform of the U.S. healthcare system, some of which could further limit coverage
and reimbursement of drug products, including combined company’s product candidates. Any reduction in reimbursement from Medicare
or other government programs may result in a similar reduction in payments from private payors. The combined company’s results of
operations could be adversely affected by the Affordable Care Act and by other health care reforms that may be enacted or adopted in the
future. In addition, the delivery of healthcare in the European Union, including the establishment and operation of health services, is
almost exclusively a matter for national, rather than EU, law and policy. National governments and health service providers have different
priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. Coupled with ever-increasing
EU and national regulatory burdens on those wishing to develop and market products, this could prevent or delay additional marketing approval
of product candidates or any initial marketing approval for combined company’s product candidates, restrict or regulate post-approval
activities and affect combined company’s ability to commercialize any products for which the combined company may obtain marketing
approval.
NLS and Kadimastem are currently
unable to predict what additional legislation or regulation, if any, relating to the health care industry may be enacted in the future
or what effect recently enacted federal legislation or any such additional legislation or regulation would have on combined company’s
business. The pendency or approval of such proposals or reforms could result in a decrease in the price of combined company’s capital
stock or limit the combined company’s ability to raise capital or to enter into collaboration agreements for the further development
and potential commercialization of combined company’s products.
Inadequate funding for the FDA and other
government agencies and/or potentially shifting priorities under the new administration could hinder the FDA’s and/or those other
government agencies’ ability to hire and retain key leadership and other personnel, prevent new products and services from being
developed or commercialized in a timely manner, or otherwise prevent those agencies from performing normal business functions on which
the operation of the combined company’s business may rely, which could negatively impact its business.
The ability of the FDA to
review and approve new products, provide feedback on clinical trials and development programs, meet with sponsors and otherwise review
regulatory submissions can be affected by a variety of factors, including government budget and funding levels; ability to hire and retain
key personnel and accept the payment of user fees; and statutory, regulatory, and policy changes, among other factors. Average review
times at the agency may fluctuate as a result. In addition, government funding of other government agencies on which the combined company’s
operations may rely is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and
other agencies may also increase the time necessary for new drugs to be reviewed and/or approved by necessary government agencies or to
otherwise respond to regulatory submissions, which would adversely affect its business. For example, the Trump Administration
has discussed several changes to the reach and oversight of the FDA, which could affect its relationship with the pharmaceutical industry,
transparency in decision making and ultimately the cost and availability of prescription drugs. Additionally, over the last several years,
the U.S. government has shut down multiple times and certain regulatory agencies, such as the FDA, have had to furlough critical
FDA and other government employees and stop critical activities. If funding for the FDA is reduced, FDA priorities change, or a prolonged
government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process combined company’s
regulatory submissions, which could have a material adverse effect on its business.
Kadimastem will need to obtain FDA approval
of any proposed names for its drug substances that gain marketing approval, and any failure or delay associated with such naming approval
may adversely impact its business.
Any name Kadimastem
intends to use for its drug substances and product candidates will require approval from the FDA regardless of whether Kadimastem
has secured a formal trademark registration from the U.S. Patent and Trademark Office, or the U.S. PTO. The FDA
typically conducts a review of proposed product names, including an evaluation of whether proposed names may be confused with the
names of other medical products and technology. The FDA may object to any product name it submits if it believes the name
inappropriately implies medical claims. If the FDA objects to any of its proposed product names, Kadimastem may be required to adopt
an alternative name for its product candidates, which could result in further evaluation of proposed names with the potential for
additional delays and costs.
Kadimastem may seek designations
for its drug substances and product candidates with the FDA and other comparable regulatory authorities that are intended to confer benefits
such as a faster development process or an accelerated regulatory pathway, but there can be no assurance that Kadimastem will successfully
obtain such designations. In addition, even if one or more of Kadimastem’s drug substances and product candidates are granted such
designations, Kadimastem may not be able to realize the intended benefits of such designations.
The FDA, and other comparable
regulatory authorities, offer certain designations for drug substances and product candidates that are intended to encourage the research
and development of pharmaceutical products addressing conditions with significant unmet medical need. These designations may confer benefits
such as additional interaction with regulatory authorities, a potentially accelerated regulatory pathway and priority review. There can
be no assurance that Kadimastem will successfully obtain such designation for Kadimastem’s products. In addition, while such designations
could expedite the development or approval process, they generally do not change the standards for approval. Even if Kadimastem obtains
such designations for one or more of its product candidates, there can be no assurance that it will realize their intended benefits.
For example, Kadimastem may
seek a Breakthrough Therapy designation from the FDA for one or more of its product candidates. A Breakthrough Therapy designation is
defined as a therapy that is intended, alone or in combination with one or more other therapies, to treat a serious or life-threatening
disease or condition, if preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing
therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.
For therapies that have Breakthrough Therapy designation, interaction and communication between the FDA and the sponsor of the trial can
help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control
regimens. Therapies with Breakthrough Therapy designation from the FDA are also eligible for accelerated approval. Designation as a Breakthrough
Therapy is within the discretion of the FDA. Accordingly, even if Kadimastem believes one of Kadimastem’s drug substances and
product candidates meets the criteria for Breakthrough Therapy designation, the FDA may disagree and instead determine not to make such
designation. In any event, the receipt of a Breakthrough Therapy designation for a product candidate may not result in a faster development
process, review or approval compared to therapies considered for approval under conventional FDA procedures and does not assure ultimate
approval by the FDA. In addition, even if one or more of Kadimastem’s drug substances and product candidates qualify for Breakthrough
Therapy designation, the FDA may later decide that such drug substances and product candidates no longer meet the conditions for qualification.
Kadimastem may also seek Regenerative
Medicine Advance Therapy, or RMAT, designation from the FDA for some of its product candidates. As described in Section 3033 of the
21st Century Cures Act, a drug is eligible for RMAT designation if: the drug is a regenerative medicine therapy, which is defined
as a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies
or products, except for those regulated solely under Section 361 of the Public Health Service Act and part 1271 of Title 21, Code
of Federal Regulations; the drug is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and
preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such disease or condition.
The FDA has broad discretion whether or not to grant this designation, so even if Kadimastem believes a particular product candidate is
eligible for this designation, there can be no assurance that the FDA would decide to grant it. Even if Kadimastem does receive RMAT designation,
Kadimastem may not experience a faster development process, review or approval compared to conventional FDA procedures, and receiving
a RMAT Designation does not provide assurance of ultimate FDA approval. The FDA may withdraw RMAT designation if it believes that the
designation is no longer supported by data from its clinical development program.
Kadimastem may also seek
Fast Track designation from the FDA for some of its product candidates. If a therapy is intended for the treatment of a serious or
life-threatening condition and the therapy demonstrates the potential to address unmet medical needs for this condition, the therapy
sponsor may apply for Fast Track designation. The FDA has broad discretion whether or not to grant this designation, so even if it
believes a particular product candidate is eligible for this designation, there can be no assurance that the FDA would decide to
grant it. Even if Kadimastem does receive Fast Track designation, it may not experience a faster development process, review or
approval compared to conventional FDA procedures, and receiving a Fast Track Designation does not provide assurance of ultimate FDA
approval. The FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data from its
clinical development program.
If Kadimastem’s manufacturing, research
and development and operational facilities are damaged or destroyed, its business and prospects would be adversely affected.
If its manufacturing, research
and development and operational facilities, or the equipment in such facilities were to be damaged or destroyed, the loss of some or all
of the stored units of its cell therapy drug substances would force Kadimastem to delay or halt its clinical trial processes. Kadimastem
has a clinical research and development facility located in Ness-Ziona, Israel, and also collaborates with medical hubs in California
and other locations in the United States. If these facilities or the equipment in them are significantly damaged or destroyed, Kadimastem
may not be able to quickly or inexpensively replace its manufacturing research and development and operational capacity.
Kadimastem’s product development is
based on novel technologies and are inherently risky.
Kadimastem is subject to the risks of failure
inherent in the development of products based on new technologies. The novel nature of its therapeutics creates significant challenges
in regard to product development and optimization, manufacturing, government regulation, third party reimbursement and market acceptance.
For example, the FDA, the EMA and other countries’ regulatory authorities have relatively limited experience with cell therapies.
Very few cell therapy products have been approved by regulatory authorities to date for commercial sale, and the pathway to regulatory
approval for its cell therapy drug substances and product candidates may accordingly be more complex and lengthier. As a result, the development
and commercialization pathway for its therapies may be subject to increased uncertainty, as compared to the pathway for new conventional
drugs.
Kadimastem’s cell therapy drug candidates
represent new classes of therapy that the marketplace may not understand or accept.
Even if Kadimastem successfully
develops and obtain regulatory approval for Kadimastem’s cell therapy candidates, the market may not understand or accept them.
Kadimastem is developing cell therapy drug substances and product candidates that represent novel treatments and will compete with a number
of more conventional products and therapies manufactured and marketed by others, including major pharmaceutical companies. The degree
of market acceptance of any of its developed and potential products will depend on a number of factors, including:
| ● | the clinical safety and effectiveness of Kadimastem’s
cell therapy drug candidates and their perceived advantage over alternative treatment methods, if any; |
| ● | adverse events involving Kadimastem’s cell therapy product
candidates or the products or product candidates of others that are cell-based; and |
| ● | the cost of Kadimastem’s products and the reimbursement
policies of government and private third party payers |
If the health care community
does not accept Kadimastem’s potential products for any of the foregoing reasons, or for any other reason, it could affect its sales,
having a material adverse effect on Kadimastem’s business, financial condition and results of operations.
Even if Kadimastem obtains regulatory approval
for a product candidate, its products will remain subject to ongoing regulatory oversight.
Even if Kadimastem
obtains any regulatory approval for its product candidates, they will be subject to ongoing regulatory requirements for
manufacturing (including manufacturing sites), labeling, packaging, storage, advertising, promotion, sampling, record-keeping and
submission of safety and other post-market information. Any regulatory approvals that it receives for its product candidates also
may be subject to a REMS, limitations on the approved indicated uses for which the product may be marketed or to the conditions of
approval or contain requirements for potentially costly post-marketing manufacturing site improvements and testing, including
Phase 4 clinical trials, and surveillance to monitor the quality, safety and efficacy of the product. The holder of an approved
BLA also must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product,
manufacturing facility, product labeling or manufacturing process. Advertising and promotional materials must comply with FDA rules
and are subject to FDA review, in addition to other potentially applicable federal and state laws.
In addition, product manufacturers
and their facilities are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory
authorities for compliance with current good manufacturing practices, or cGMP, requirements and adherence to commitments made in the BLA
or foreign marketing application. If Kadimastem, or a regulatory authority, discover previously unknown problems with a product, such
as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured or disagrees
with the promotion, marketing or labeling of that product, a regulatory authority may impose restrictions relative to that product, the
manufacturing facility or Kadimastem, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If Kadimastem fails to comply
with applicable regulatory requirements, a regulatory authority may:
| ● | issue an untitled letter or warning letter that Kadimastem
is in violation of the law; |
| ● | seek an injunction or impose administrative, civil or criminal
penalties or monetary fines; |
| ● | suspend or withdraw regulatory approval; |
| ● | suspend any ongoing clinical trials; |
| ● | refuse to approve pending applications or supplements to applications; |
| ● | restrict the marketing or manufacturing of the product; |
| ● | seize or detain the products or require the withdrawal of
the product from the market; |
| ● | refuse to permit the import or export of the products; or |
| ● | refuse to allow Kadimastem to enter into supply contracts,
including government contracts. |
Any government investigation
of alleged violations of law could require it to expend significant time and resources in response and could generate negative publicity.
The occurrence of any event or penalty described above may inhibit Kadimastem’s ability to commercialize its product candidates
and adversely affect its business, financial condition, results of operations and prospects.
International expansion of Kadimastem’s
business exposes it to business, regulatory, political, operational, financial and economic risks associated with doing business outside
of the United States, within Israel, the EU and Japan.
Other than Kadimastem’s
headquarters and other operations which are located in Israel, it currently have limited international operations, but Kadimastem’s
business strategy incorporates potentially significant international expansion, particularly in anticipation of approval of its product
candidates. Kadimastem plans to maintain research and development, manufacturing, development, and sales representatives and conduct physician
and patient association outreach activities, as well as clinical trials, within and outside of the United States, in Israel, Europe.
and Japan. If Kadimastem’s products are approved for commercialization outside the United States, Israel, or the EU, Kadimastem
will likely enter into agreements with third parties to market its drug substances in these additional global territories. Kadimastem
expects that it will be subject to additional risks related to entering into or maintaining international business relationships, including:
| ● | different regulatory requirements for approvals of drug substances
in foreign countries; |
| ● | differing United States and foreign drug import and export
rules, tariffs and other trade barriers; |
| ● | reduced protection for intellectual property rights in foreign
countries; |
| ● | failure by Kadimastem to obtain regulatory approvals for the
use of its products in various countries; |
| ● | different reimbursement systems; |
| ● | economic weakness, including inflation, or political instability
in particular foreign economies and markets; |
| ● | multiple, conflicting and changing laws and regulations such
as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals,
permits and licenses; |
| ● | complexities associated with managing multiple payor reimbursement
regimes, government payors or patient self-pay systems; |
| ● | financial risks, such as longer payment cycles, difficulty
collecting accounts receivable, the impact of local and regional financial crises on demand and payment for its products and exposure
to foreign currency exchange rate fluctuations, which could result in increased operating expenses and reduced revenues; |
| ● | workforce uncertainty in countries where labor unrest is more
common than in the United States; |
| ● | production shortages resulting from any events affecting raw
material supply or manufacturing capabilities abroad; |
| ● | regulatory and compliance risks that relate to maintaining
accurate information and control over sales and activities that may fall within the purview of the U.S. Foreign Corrupt Practices
Act, its books and records provisions or its anti-bribery provisions; |
| ● | potential liability resulting from development work conducted
by these distributors; and |
| ● | business interruptions resulting from a local or worldwide
pandemic, such as geopolitical actions, including war and terrorism, or natural disasters. |
Any of these factors could
significantly harm its future international expansion and operations and, consequently, Kadimastem’s results of operations.
Even if any of Kadimastem’s product
candidates receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors
and others in the medical community necessary for commercial success.
The commercial success of
Kadimastem’s products will depend upon the acceptance of each product by the medical community, including physicians, patients and
third-party payors. The degree of market acceptance of any approved product will depend on a number of factors, including:
| ● | the efficacy and safety of the product; |
| ● | the potential advantages of the product compared to available
therapies; |
| ● | the convenience and ease of administration compared to alternative
treatments; |
| ● | limitations or warnings, including use restrictions contained
in the product’s approved labeling; |
| ● | distribution and use restrictions imposed by the EMA, FDA
or other regulatory authority or agreed to by Kadimastem as part of a mandatory or voluntary risk management plan; |
| ● | pricing and cost effectiveness in relation to alternative
treatments; |
| ● | if the product is included under physician treatment guidelines
as a first-, second,- or third-line therapy; |
| ● | the strength of sales, marketing and distribution support; |
| ● | the availability of third-party coverage and adequate reimbursement
and the willingness of patients to pay out-of-pocket in the absence of coverage by third-party payors; |
| ● | the strength of sales, marketing and distribution support; |
| ● | the willingness of patients to pay for drugs out of pocket
in the absence of third-party coverage; and |
| ● | the willingness of the target patient population to try new
therapies and of physicians to prescribe these therapies. |
If Kadimastem’s products
are approved but do not achieve an adequate level of acceptance by physicians, third party payors and patients, Kadimastem may not generate
sufficient revenue from the product, and Kadimastem may not become or remain profitable. In addition, its efforts to educate the medical
community and third-party payors on the benefits of the product may require significant resources and may never be successful.
In addition, Kadimastem
may choose to collaborate with third parties that have direct sales forces and established distribution systems, either to augment
its own sales force and distribution systems or in lieu of its own sales force and distribution systems. If Kadimastem enters into
arrangements with third parties to perform sales, marketing and distribution services for its products, the resulting revenues or
the profitability from these revenues to it are likely to be lower than if Kadimastem had sold, marketed and distributed its
products itself. If Kadimastem is unable to enter into such arrangements on acceptable terms or at all, Kadimastem may not be able
to successfully commercialize any of Kadimastem’s product candidates that receive regulatory approval. Depending on the nature
of the third-party relationship, Kadimastem may have little control over such third parties, and any of these third parties may fail
to devote the necessary resources and attention to sell, market and distribute its products effectively. If Kadimastem is not
successful in commercializing its product candidates, either on Kadimastem’s own or through collaborations with one or more
third parties, its future product revenue will suffer and Kadimastem may incur significant additional losses.
Even if Kadimastem is able to commercialize
any product candidates, the products may become subject to unfavorable pricing regulations or third-party coverage and reimbursement policies,
any of which could harm its business.
Kadimastem’s ability
to commercialize any product candidates successfully will depend, in part, on the extent to which coverage and reimbursement for these
products and related treatments will be available from government health administration authorities, private health insurers and other
organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide
which medications they will pay for and impact reimbursement levels.
Obtaining and maintaining
adequate reimbursement for Kadimastem’s products may be difficult. Kadimastem cannot be certain if and when Kadimastem will obtain
an adequate level of reimbursement for its products by third party payors. Even if it does obtain adequate levels of reimbursement, third-party
payors, such as government or private healthcare insurers, carefully review and increasingly question the coverage of, and challenge the
prices charged for, drugs. Reimbursement rates from private health insurance companies vary depending on the company, the insurance plan
and other factors. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and
third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly,
third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the
prices charged for drugs. Kadimastem may also be required to conduct expensive pharmacoeconomic studies to justify coverage and reimbursement
or the level of reimbursement relative to other therapies. If coverage and reimbursement are not available or reimbursement is available
only to limited levels, Kadimastem may not be able to successfully commercialize any product candidate for which Kadimastem obtain marketing
approval, and the royalties resulting from the sales of those products may also be adversely impacted.
There may be significant
delays in obtaining reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is
approved by the FDA or similar regulatory authorities outside the United States. Moreover, eligibility for reimbursement does
not imply that a drug will be paid for in all cases or at a rate that covers Kadimastem’s costs, including research,
development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be
sufficient to cover Kadimastem’s costs and may not be made permanent. Reimbursement rates may vary according to the use of the
drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be
incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates
required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of
drugs from countries where they may be sold at lower prices than in the United States. Kadimastem’s inability to promptly
obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products that it
develops could have a material adverse effect on its operating results, its ability to raise capital needed to commercialize
products and its overall financial condition.
The regulations that govern
marketing approvals, pricing, coverage, and reimbursement for new drug substances vary widely from country to country. Current and future
legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining
approvals. Some countries require approval of the sale price of drug substances before they can be reimbursed. In many countries, the
pricing review period begins after marketing or drug substances licensing approval is granted. In some foreign markets, prescription drug
substances pricing remains subject to continuing governmental control, including possible price reductions, even after initial approval
is granted. As a result, Kadimastem might obtain marketing approval for drug substances in a particular country, but then be subject to
price regulations that delay Kadimastem’s commercial launch of its products and technology, possibly for lengthy time periods, and
negatively impact the revenues it is able to generate from the sale of drug substances in that country. Adverse pricing limitations may
hinder Kadimastem’s ability to recoup its investment in one or more products and technology, even if its drug substances obtain
marketing approvals. There can be no assurance that Kadimastem’s products and technology, if they are approved for sale in the United States
or in other countries, will be considered medically necessary or cost-effective for a specific indication, or that coverage or an adequate
level of reimbursement will be available.
The FDA and other regulatory authorities
actively enforce the laws and regulations prohibiting the promotion of off-label uses.
If any of Kadimastem’s
drug substances are approved and it is found to have improperly promoted off-label uses of those products and technology, Kadimastem may
become subject to significant liability. The FDA and other regulatory agencies strictly regulate the promotional claims that may be made
about prescription of its products and technology, if approved. If Kadimastem is found to have promoted such off-label uses, it may become
subject to significant liability. The federal government has levied large civil and criminal fines against companies for alleged improper
promotion of off-label use and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies
enter into consent decrees, corporate integrity agreements or permanent injunctions under which specified promotional conduct must be
changed or curtailed. If Kadimastem cannot successfully manage the promotion of its products and technology, if approved, Kadimastem could
become subject to significant liability, which would materially adversely affect Kadimastem’s business and financial condition.
Kadimastem may be subject, directly or indirectly,
to federal and state healthcare fraud and abuse laws, false claims laws and health information privacy and security laws. If Kadimastem
is unable to comply, or have not fully complied, with such laws, it could face substantial penalties.
If Kadimastem obtains FDA
approval for any of its product candidates and begin commercializing those products in the United States, its operations may be directly
or indirectly through its customers, subject to various federal and state fraud and abuse laws, including, without limitation, the federal
Anti-Kickback Statute, the federal False Claims Act and physician sunshine laws and regulations. These laws may impact, among other things,
its proposed sales, marketing and education programs. In addition, Kadimastem may be subject to patient privacy regulation by both the
federal government and the states in which it conducts its business. The laws that may affect its ability to operate include:
| ● | the federal Anti-Kickback Statute, which prohibits, among
other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to
induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such
as the Medicare and Medicaid programs; |
| ● | federal civil and criminal false claims laws and civil monetary
penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims
for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent; |
| ● | the Health Insurance Portability and Accountability Act of 1996,
or HIPAA, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and
making false statements relating to healthcare matters; |
| ● | HIPAA, as amended by the Health Information Technology and
Clinical Health Act, and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission
of individually identifiable health information; |
| ● | the federal physician sunshine requirements under the Patient
Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, requires manufacturers of drugs,
devices and medical supplies to report annually to the U.S. Department of Health and Human Services information related to payments
and other transfers of value to physicians, other healthcare providers and teaching hospitals and ownership and investment interests
held by physicians and other healthcare providers and their immediate family members and applicable group purchasing organizations; |
| ● | state law equivalents of each of the above federal laws, such
as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial
insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines
and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare
providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments
and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy
and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have
the same effect, thus complicating compliance efforts; and |
| ● | European and other foreign law equivalents of each of the
laws, including reporting requirements detailing interactions with and payments to healthcare providers, and the European General Data
Protection Regulation, or GDPR, which contains provisions specifically directed at the processing of health information, higher sanctions
and extra-territoriality measures intended to bring non-EU companies under the regulation, including companies like it that conduct clinical
trials in the EU; Kadimastem anticipates that over time it may expand its business operations to include additional operations in the
EU and with such expansion, it would be subject to increased governmental regulation in the EU countries in which it might operate, including
the GDPR. |
The scope and enforcement
of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of
the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions
between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements
in the healthcare industry. It is possible that governmental authorities will conclude that Kadimastem’s business practices do not
comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations.
If Kadimastem’s operations are found to be in violation of any of these laws or any other related governmental regulations that
may apply to it, Kadimastem may be subject to significant civil, criminal and administrative penalties, including, without limitation,
damages, fines, imprisonment, disgorgement, exclusion from participation in government funded healthcare programs, such as Medicare and
Medicaid, reputational harm, additional oversight and reporting obligations if Kadimastem becomes subject to a corporate integrity agreement
or similar settlement to resolve allegations of non-compliance with these laws and the curtailment or restructuring of Kadimastem’s
operations. If any of the physicians or other healthcare providers or entities with whom Kadimastem expects to do business is found to
be not in compliance with applicable laws, they may be subject to similar actions, penalties and sanctions. Efforts to ensure that its
business arrangements comply with applicable healthcare laws and regulations, as well as responding to possible investigations by government
authorities, can be time- and resource-consuming and can divert a company’s attention from the business.
Obtaining and maintaining
regulatory approval of Kadimastem’s drug substances in one jurisdiction does not mean that it will be successful in obtaining regulatory
approval of Kadimastem’s drug substances in other jurisdictions. Kadimastem’s failure to obtain regulatory approval in foreign
jurisdictions would prevent its drug substances from being marketed abroad, and any approval it is granted for Kadimastem’s drug
substances in the United States would not assure approval of drug substances in foreign jurisdictions.
In order to market any
products outside of the United States, Kadimastem must establish and comply with numerous and varying regulatory requirements
of other countries regarding clinical trial design, safety and efficacy. The research, testing, manufacturing, labeling, approval,
sale, marketing and distribution of drugs are subject to extensive regulation by the FDA in the United States and other
regulatory authorities in other countries. These regulations differ from country to country. Even if Kadimastem obtains and
maintains regulatory approval of its product candidates in one jurisdiction, such approval does not guarantee that Kadimastem will
be able to obtain or maintain regulatory approval in any other jurisdiction, but a failure or delay in obtaining regulatory approval
in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants
marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the
manufacturing, marketing and promotion of the product candidate in those countries.
Approval procedures vary among
jurisdictions and can involve requirements and administrative review periods different from those in the United States, including
additional nonclinical studies or clinical trials as investigations conducted in one jurisdiction may not be accepted by regulatory authorities
in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before
it can be approved for sale in that jurisdiction. In some cases, the price that Kadimastem intends to charge for its products is also
subject to approval. These regulatory procedures can result in substantial delays in such countries. In other countries, product approval
depends on showing superiority to an approved alternative therapy. This can result in significant expense for conducting complex clinical
trials.
If Kadimastem, or any third
parties with whom it works, fail to comply with regulatory requirements in the United States or international markets, or fail to
obtain and maintain required approvals, or if regulatory approvals in international markets are delayed, Kadimastem’s target market
may be reduced and its ability to realize the full market potential of its products will likely be harmed. The inability to meet continuously
evolving regulatory standards for approval may result in its failing to obtain regulatory approval to market Kadimastem’s current
product candidates, which could significantly harm its business, results of operations and prospects.
Kadimastem’s market is subject to
intense competition, which may result in others commercializing products before or more successfully. If Kadimastem is unable to compete
effectively, its products may be rendered noncompetitive or obsolete, which may adversely affect its operating results.
The development and commercialization
of new products is highly competitive. Kadimastem’s potential competitors include major pharmaceutical companies, specialty pharmaceutical
companies and biotechnology companies worldwide with respect to its products or any future product candidate that Kadimastem may seek
to develop or commercialize. Kadimastem’s competitors may succeed in developing, acquiring or licensing technologies and products
that are more effective, have fewer or more tolerable side effects or are more convenient or less costly than its products or any future
product candidate it may develop, which could render any product candidates obsolete and noncompetitive. Kadimastem’s competitors
also may obtain FDA or other marketing approval for their products before it is able to obtain approval for Kadimastem’s, which
could result in competitors establishing a strong market position before it is able to enter the applicable market.
Many of Kadimastem’s
potential competitors, alone or with their strategic partners, have significantly greater financial resources and expertise in research
and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining marketing approvals and commercializing approved
products than it does. There is a trend toward consolidation in the pharmaceutical and biotechnology industry, and additional mergers
and acquisitions in these industries may result in even more resources being concentrated among a smaller number of Kadimastem’s
competitors, which may adversely affect it.
Smaller or early-stage companies
may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These
companies also compete with Kadimastem in recruiting and retaining qualified scientific and management personnel and establishing clinical
trial sites and patient registration for clinical trials.
In addition, if Kadimastem
enters the markets of its product candidates, with such entrance remaining subject to various additional regulatory approvals, too late
in the cycle, Kadimastem may not achieve commercial success, or it may have to reduce its price in order to effectively compete, which
would impact its ability to generate revenue, obtain profitability and adversely affect its operating results.
The major market players within
the ALS and diabetes drug markets and Kadimastem’s primary competitors in the United States and abroad include among others
Biogen and Mitsubishi Tanabe pharmaceuticals in the ALS domain and Vertex Therapeutics and Novo-Nordisk pharmaceuticals in the cell therapy
for diabetes domain. Some of these companies hold significant market share. Their dominant market position and significant control over
the market could significantly limit its ability to introduce or effectively market and generate sales and capture market share.
Competition in the pharmaceutical
industry is intense, and can lead to, among other things, price reductions, longer selling cycles, lower product margins, loss of market
share, and additional working capital requirements. To succeed, Kadimastem must, among other critical matters, gain consumer acceptance
for its products, as compared to other solutions currently available in the market. For example, since the currently accepted treatment
for ALS is not substantially effective for its patients and for diabetes is lacking in effectiveness and does not cure the disease itself
nor does it prevent long term complications, respectively, Kadimastem will need to invest resources in educating the medical community
and consumers, and establish strategic collaborations before Kadimastem will be able to gain market acceptance for Kadimastem’s
AstroRx® and/or IsletRx as a treatment in severe acute emergency situations. If Kadimastem’s competitors offer significant
discounts on certain products and solutions, Kadimastem may need to lower its prices or offer other favorable terms in order to compete
successfully. Moreover, any broad-based changes to Kadimastem’s prices and pricing policies could make it difficult to generate
revenues or cause Kadimastem’s revenues to decline. Moreover, if Kadimastem’s competitors develop and commercialize products
and solutions that are more effective or desirable than products and solutions that it may develop, Kadimastem may not convince its customers
to use its products and solutions. Any such changes would likely reduce Kadimastem’s commercial opportunity and revenues potential
and could materially adversely impact Kadimastem’s operating results.
Although Kadimastem has obtained orphan
drug designation from the EMA for its product AstroRx®, it may not be able to realize the benefits of such designation,
including potential marketing exclusivity of its product candidates, if approved.
Kadimastem applied for and
received designation from the FDA for AstroRx® for cell therapy for ALS treatment. Regulatory authorities in some jurisdictions,
including the EU and United States, may designate drugs for relatively small patient populations as “orphan drugs.” In
the EU, the European Commission grants orphan drug designation to promote the development of products that are intended for the diagnosis,
prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in
the EU community. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening,
seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the EU
would be sufficient to justify the necessary investment in developing the drug. In the United States, under the Orphan Drug Act the
FDA may designate a product as an orphan drug if it is a drug intended to treat a rare disease or condition, which is generally defined
as a patient population of fewer than 200,000 individuals in the United States or a patient population of greater than 200,000 individuals
in the United States, but for which there is no reasonable expectation that the cost of developing the drug will be recovered from
sales in the United States
The designation of a product
candidate as an orphan product does not mean that any regulatory agency will accelerate regulatory review of, or ultimately approve, that
product candidate, nor does it limit the ability of any regulatory agency to grant orphan drug designation to product candidates of other
companies that treat the same indications as its product candidates prior to Kadimastem’s product candidates receiving exclusive
marketing approval.
Generally, if a product candidate
with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation,
the drug may be entitled to a period of marketing exclusivity, which precludes the FDA or the EMA from approving another marketing application
for the same drug for that time period, except in limited circumstances.
Although Kadimastem has obtained
orphan drug exclusivity from the FDA for AstroRx®, that exclusivity may not effectively protect the product from competition
because different drugs can be approved for the same condition. Even after an orphan drug is approved, the FDA may subsequently approve
another drug for the same condition if the FDA concludes that the latter drug is not the same drug or is clinically superior in that it
is shown to be safer, more effective or makes a major contribution to patient care. In the EU, marketing authorization may be granted
to a similar medicinal product for the same orphan indication if:
| ● | the second applicant can establish in its application that
its medicinal product, although similar to the orphan medicinal product already authorized, is safer, more effective or otherwise clinically
superior; |
| ● | the holder of the marketing authorization for the original
orphan medicinal product consents to a second orphan medicinal product application; or |
| ● | the holder of the marketing authorization for the original
orphan medicinal product cannot supply sufficient quantities of orphan medicinal product. |
Sales of Kadimastem’s
approved products, if any, will be subject to the regulatory requirements governing marketing approval in the countries in which Kadimastem
obtains regulatory approval, and where it plans to seek itself or with collaborators regulatory approval to commercialize its product
candidates in North America, the EU and in additional foreign countries. Clinical trials conducted in one country may not be accepted
by regulatory authorities in other countries and regulatory approval in one country does not ensure approval in any other country, while
a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory approval process in others.
For example, approval in the U.S. by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions,
and approval by one foreign regulatory authority does not ensure approval by the FDA, EMA, or regulatory authorities in other countries.
Approval procedures vary among jurisdictions and can be lengthy and expensive, and involve requirements and administrative review periods
different from, and potentially greater than, those in the U.S., including additional pre-clinical studies or clinical trials. Even if
Kadimastem’s product candidates are approved, regulatory approval for any product may be withdrawn by the regulatory authorities
in a particular jurisdiction.
Even if a product is approved,
the FDA, EMA or another applicable regulatory authority, as the case may be, may limit the indications for which the product may be marketed,
require extensive warnings on the product labeling or require expensive and time-consuming post-approval commitments including clinical
trials or onerous risk management activities, including REMS, in the U.S. as conditions of approval to help ensure that the benefits
of the drug outweigh the potential risks. REMS can include medication guides, communication plans for health care professionals, and elements
to assure safe use. Elements to assure safe use can include, but are not limited to, special training or certification for prescribing
or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for
a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval
testing and surveillance to monitor the drug’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance
with regulatory standards is not maintained or problems are identified following initial marketing. In many countries outside the U.S.,
a product candidate must be approved for reimbursement before it can be approved for sale in that country. In some cases, the price that
Kadimastem intend to charge for a product is also subject to approval.
Regulatory authorities in
countries outside of the U.S. and the EU also have their own requirements for approval of product candidates with which Kadimastem
must comply prior to marketing in those countries. Obtaining foreign regulatory approvals and compliance with such foreign regulatory
requirements could result in significant delays, difficulties and costs for Kadimastem or its collaborators and could delay or prevent
the introduction of its current and any future products, in certain countries.
If Kadimastem or its collaborators
fail to receive applicable marketing approvals or comply with the regulatory requirements in international markets, its target market
will be reduced and its ability to realize the full market potential of its product candidates will be harmed and its business will be
adversely affected.
If product liability lawsuits are brought
against Kadimastem, it may incur substantial liabilities, even if Kadimastem has appropriate insurance policies, and it may be required
to limit commercialization of its product candidates.
Kadimastem is exposed to
potential product liability and professional indemnity risks that are inherent in the research, development, manufacturing,
marketing and use of pharmaceutical products. Currently, it has no products that have been approved for marketing or
commercialization; however, the use of its product candidates in clinical trials, and the sale of these product candidates, if
approved, in the future, may expose it to liability claims. Product liability claims may be brought against Kadimastem or its
partners by participants enrolled in its clinical trials, patients, health care providers, pharmaceutical companies, its
collaborators or others using, administering or selling any of its future approved products. If Kadimastem cannot successfully
defend itself against any such claims, it may incur substantial liabilities, even if Kadimastem has product liability or such other
applicable insurance policies in effect. Kadimastem may not be able to maintain adequate levels of insurance for these liabilities
at reasonable cost and/or reasonable terms.
Excessive insurance costs
or uninsured claims would add to its future operating expenses and adversely affect its financial condition. As a result of such lawsuits
and their potential results, it may be required to limit commercialization of its product candidates. Regardless of the merits or eventual
outcome, liability claims may result in:
| ● | decreased demand for its drug substances and product candidates; |
| ● | termination of clinical trial sites or entire trial programs; |
| ● | injury to its reputation and negative media attention; |
| ● | product recalls or increased warnings on product labels; |
| ● | withdrawal of clinical trial participants; |
| ● | costs of to defend the related litigation; |
| ● | diversion of management and its resources; |
| ● | substantial monetary awards to, or costly settlements with,
clinical trial participants, patients or other claimants; |
| ● | higher insurance premiums; |
| ● | loss of initiation of investigations by regulators or other
authorities; and |
| ● | the inability to successfully commercialize its drug substances
and product candidates, if approved. |
Kadimastem’s inability
to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims
could prevent or inhibit the commercialization of products it develops or acquires. Kadimastem intends to obtain product liability insurance
covering its clinical trials. Although Kadimastem will maintain such insurance, any claim that may be brought against Kadimastem could
result in a court judgment or settlement in an amount that is not covered, in whole or in part, by its insurance or that is in excess
of the limits of Kadimastem’s insurance coverage. Kadimastem’s insurance policies also have various exclusions, and it may
be subject to a product liability claim for which Kadimastem has no coverage. Kadimastem may have to pay any amounts awarded by a court
or negotiated in a settlement that exceed its coverage limitations or that are not covered by its insurance, and Kadimastem may not have,
or be able to obtain, sufficient capital to pay such amounts.
Risks Related to the Combined Company’s
Business and Industry
The operations and commercialization of
stem cell therapies is a new and integral part of the emerging regenerative medicine market, but the field remains in its infancy.
As with all new technologies,
products, practices and solutions, there are inherit risks related to combined company’s industry and business.
The field of stem cell therapy
is relatively new, and not yet widely adopted by the medical community, and because of that infancy, it may have an adverse effect on
its ability to reach potential physicians that are skeptical of the benefits or have questions about the risks, and thus, the combined
company may run into resistance in the marketing of its products and services. Stem cell therapies may be susceptible to various risks,
including side effects, unintended immune system responses, inadequate therapeutic efficacy, and lack of acceptance by physicians, hospital,
and the patients themselves.
The combined company’s
experience and others have shown that physicians are historically slow to adopt new treatment methods based on new technologies, like
the combined company’s, when existing and trusted methods continue to be supported by established practitioners. Overcoming these
obstacles often requires significant marketing expenditures, product performance, cost cutting and/or decreased pricing. It believes the
skepticism to be a significant barrier as the combined company’s attempts to gain market penetration with its products and services.
Failure to achieve market acceptance of its products and services would have a material adverse effect on the combined company’s
financial condition.
Additionally, part of the
combined company’s success will depend on continuing to establish and maintain effective strategic partnerships and collaborations
with its international partners, which may impose challenges, restrictions, and or financial impacts to its business.
As the combined company applies
its business strategy of establishing and maintaining strategic relationships, it believes this will allow the combined company to expand
and complement its products, training, support and commercialization capabilities. This the combined company believes will allow it to
reduce costs with greater economies of scale, and leverage a greater source of market intelligence, with crucial meta data gathered of
stem cell therapies applied to a full spectrum across global applications. Notwithstanding, there can be no assurances that the combined
company will favorably maintain all current or successfully add new relationships to successfully advance its business.
The combined company’s
likelihood of profitability depends on its ability to license and/or develop and commercialize its products based on its cell production
technology, which is currently in the development stage. If the combined company is unable to complete the development and commercialization
of its cell therapy products successfully, its likelihood of profitability will be limited severely.
The combined company is engaged
in the business of developing cell therapy products. It has not realized a profit from its operations to date and there is little likelihood
that the combined company will realize any profits in the short or medium term. Any profitability in the future from its business will
be dependent upon successful commercialization of its potential cell therapy products and/or licensing of its products, which will require
additional research and development.
The clinical manufacturing
process for cell therapy products is complex and requires meeting high regulatory standards. Any delay or problem in the clinical manufacturing
of its products may result in a material adverse effect on its business.
The clinical manufacturing
process is complex, and the combined company has no experience in manufacturing the combined company’s product candidates at a commercial
level. There can be no guarantee that it will be able to successfully develop and manufacture its product candidates in a manner that
is cost-effective or commercially viable, or that its development and manufacturing capabilities might not take much longer than currently
anticipated to be ready for the market. In addition, if the combined company fails to maintain regulatory approvals for its manufacturing
facilities, it may suffer delays in its ability to manufacture it product candidates. This may result in a material adverse effect on
its business.
If the combined company encounters problems
or delays in the research and development of its potential cell therapy products, it may not be able to raise sufficient capital to finance
its operations during the period required to resolve such problems or delays.
The combined
company’s cell therapy products are currently in the development stage and it anticipate that it will continue to incur
substantial operating expenses and incur net losses until it has successfully completed all necessary research and clinical trials.
The combined company, and any of its potential collaborators, may encounter problems and delays relating to research and
development, regulatory approval and intellectual property rights of its technology. The combined company’s research and
development programs may not be successful, and its cell culture technology may not facilitate the production of cells outside the
human body with the expected result. The combined company’s cell therapy products may not prove to be safe and efficacious in
clinical trials. If any of these events occur, the combined company may not have adequate resources to continue operations for the
period required to resolve the issue delaying commercialization and it may not be able to raise capital to finance its continued
operation during the period required for resolution of that issue. Accordingly, the combined company may be forced to discontinue or
suspend its operations.
Any cell-based products that receive regulatory
approval may be difficult and expensive to manufacture profitably.
Cell-based products are among
the more expensive biologic products to manufacture. The combined company does not yet have sufficient information to reliably estimate
the cost of commercially manufacturing any of its product candidates. Excessive manufacturing costs could make its product candidates
too expensive to compete in the medical marketplace with alternative products manufactured by its competitors or might result in third
party payors such as health insurers and Medicare, declining to cover its products or setting reimbursement levels too low for the combined
company to earn a profit from the commercialization of one or more of its products.
The combined company’s future success
depends on its ability to retain key executives and to attract, retain and motivate qualified personnel.
The combined company’s
future success depends to a large extent on the continued services of members of Kadimastem’s current management including, in particular,
Professor Michel Ravel, Chief Scientific Officer, and Dr. Kfir Molekandov, Kadimastem’s Vice President of Research and Development,
Dr. Ariel Revel, Director of Medical Affairs, and Ronen Twito, Kadimastem’s Executive Chairman and CEO. Any of the combined company’s
employees and consultants may leave the company at any time, subject to certain notice periods. The loss of the services of any of the
combined company’s executive officers or any key employees or consultants may adversely affect its ability to execute its business
plan and harm its operating results. The combined company’s operational success will substantially depend on the continued employment
of senior executives, technical staff and other key personnel, especially given the intense competition for qualified personnel. The loss
of key personnel may have an adverse effect on the combined company’s operations and financial performance.
Recruiting and retaining qualified
scientific and clinical personnel and, if the combined company progresses the development of its product pipeline toward scaling up for
commercialization, manufacturing and sales and marketing personnel, will also be critical to the combined company’s success. The
loss of the services of its executive officers or other key employees could impede the achievement of its development and commercialization
objectives and seriously harm its ability to successfully implement its business strategy. Furthermore, replacing executive officers and
key employees may be difficult and may take an extended period of time because of the limited number of individuals in its industry with
the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize products. Competition
to hire from this limited pool is intense, and the combined company may be unable to hire, train, retain or motivate these key personnel
on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. It also experience
competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, the combined
company’s relies on consultants and advisors, including scientific and clinical advisors, to assist the combined company in formulating
its development and commercialization strategy. The combined company’s consultants and advisors may be employed by employers other
than the combined company and may have commitments under consulting or advisory contracts with other entities that may limit their availability
to the combined company. If the combined company is unable to continue to attract and retain high quality personnel, its ability to pursue
its growth strategy will be limited.
Certain debt agreements that the combined
company may enter into may contain restrictions that may limit its flexibility in operating its business.
Future debt agreements that
the combined company may enter into could limit its flexibility in operating its business. Documents governing such future indebtedness,
or those in connection with additional capital raises, if any, may contain numerous financial and operating covenants that limit the discretion
of management with respect to certain business matters. Restrictive covenants included in these future debt agreements may include restrictions
on, among other things, the combined company’s ability to:
| ● | create or permit to subsist any security interest over any
of its assets; |
| ● | sell, transfer or otherwise dispose of any or its receivables
on recourse terms; |
| ● | buy back its own common shares; |
| ● | incur or permit additional indebtedness; |
| ● | merge or conduct any other corporate reconstruction; and |
| ● | change the nature of its business. |
The combined company’s
ability to comply with these and other provisions of future debt agreements will depend on its future performance, which will be subject
to many factors, some of which are beyond its control. The breach of any negative covenants in these future agreements could result in
an event of default, as may be defined in such agreements, potentially leading to a default interest rate or immediate repayment of any
borrowed amounts. These restrictive covenants, which may be included in future debt agreements, and any lack of compliance by the combined
company could limit its flexibility in operating its business.
The use of any of the combined company’s
product candidates could result in product liability or similar claims that could be expensive, damage its reputation and harm its business.
The combined company’s
business exposes it to an inherent risk of potential product liability or similar claims. The pharmaceutical industry has historically
been litigious, and it faces financial exposure to product liability or similar claims if the use of any of its products were to cause
or contribute to injury or death. There is also the possibility that defects in the design or manufacture of any of its products might
necessitate a product recall. Although the combined company plans to maintain product liability insurance, the coverage limits of these
policies may not be adequate to cover future claims. In the future, the combined company may be unable to maintain product liability insurance
on acceptable terms or at reasonable costs and such insurance may not provide it with adequate coverage against potential liabilities.
A product liability claim, regardless of merit or ultimate outcome, or any product recall could result in substantial costs to the combined
company, damage to its reputation, customer dissatisfaction and frustration and a substantial diversion of management attention. A successful
claim brought against the combined company in excess of, or outside of, its insurance coverage could have a material adverse effect on
the combined company business, financial condition and results of operations.
Failure in its information technology systems,
including by cybersecurity attacks or other data security incidents, could significantly disrupt the combined company’s operations.
The combined company’s
operations depend, in part, on the continued performance of its information technology systems. The combined company’s information
technology systems are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptions. Failure of
its information technology systems could adversely affect its business, profitability and financial condition. Although the combined company
has information technology security systems, a successful cybersecurity attack or other data security incident could result in the misappropriation
and/or loss of confidential or personal information, create system interruptions, or deploy malicious software that attacks its systems.
It is possible that a cybersecurity attack might not be noticed for some period of time. The occurrence of a cybersecurity attack or incident
could result in business interruptions from the disruption of the combined company’s information technology systems, or negative
publicity resulting in reputational damage with its shareholders and other shareholders and/or increased costs to prevent, respond to
or mitigate cybersecurity events. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could
result in delays in its regulatory approval efforts and significantly increase its costs to recover or reproduce the data. In addition,
the unauthorized dissemination of sensitive personal information or proprietary or confidential information could expose the combined
company or other third parties to regulatory fines or penalties, litigation and potential liability, or otherwise harm its business.
Unsuccessful compliance with certain European
privacy regulations could have an adverse effect on the combined company’s business and reputation.
The collection and use
of personal health data in the EU is governed by the provisions of the data protection derivative and the GDPR. These
directives impose several requirements relating to the consent of the individuals to whom the personal data relates, the information
provided to the individuals, notification of data processing obligations to the competent national data protection authorities and
the security and confidentiality of the personal data. The GDPR also extends the geographical scope of EU data protection law to
non-EU entities under certain conditions, tightens existing EU data protection principles and creates new obligations for companies
and new rights for individuals. Failure to comply with the requirements of the Data Protection Directive, the GDPR, and the related
national data protection laws of the EU Member States may result in fines and other administrative penalties. The GDPR introduces
new data protection requirements in the EU and substantial fines for breaches of the data protection rules. The GDPR regulations
impose additional responsibility and liability in relation to personal data that the combined company processes and the combined
company intends to put in place additional mechanisms ensuring compliance with these and/or new data protection rules. In addition,
other jurisdictions, are currently discussing or implementing regulations similar to GDPR. Changes to these European privacy
regulations (and similar regulations in other jurisdictions) and unsuccessful compliance may be onerous and adversely affect its
business, financial condition, prospects, results of operations and reputation.
Risks Related to the Combined Company’s
Reliance on Third Parties
The combined company’s reliance on
third party expenses reimbursement related to its products by insurance companies, pursuant to eligibility criteria that may be changed,
as well as fluctuations of indemnity amounts that may impair its ability to sell its products and may affect its business outcomes which
are determined by both the Israeli government and by insurance companies.
The combined company’s
activity is influenced by a policy of health insurers regarding its eligibility to participate and cover its expenses, or reimbursement,
known in Israel as the “Healthcare Basket.” The degree of participation or coverage (full or partial) for the purchase of
products or medical treatment developed or provided by it may affect its activities and the sales potential and business outcomes for
the combined company. On the other hand, the recognition of medical insurers for the purchase of products or medical care that will be
developed or provided by the combined company, for the purpose of reimbursing expenses, is expected to increase demand for its products
and services in its target markets. The entitlement to reimbursement is determined from time to time by health insurance companies, both
private insurance companies and public insurance companies, in accordance with considerations that go beyond the scope of the combined
company’s influence. A change to the reimbursement criteria or non-confirmation of eligibility for expenses reimbursement related
to its products, as well as fluctuations of indemnity amounts for substitute and complementary diagnostic tests may assist or impair its
ability to sell the combined company’s products and may affect its business outcomes.
The combined company may be materially adversely
affected if there is an imbalance in global logistics, which may cause disruptions to its transport, storage and distribution operations,
negatively impacting the costs related thereto.
The combined company’s
operations are dependent upon uninterrupted transportation, storage and distribution of its products. Transportation, storage or distribution
of its products could be partially or completely, temporarily or permanently shut down as the result of any number of circumstances that
are not within its control, such as:
| ● | strikes or other labor difficulties; |
| ● | disruption in the global supply chain, including container
shortages; |
| ● | war and other armed conflicts, such as the war in Israel and
the war involving Russia and Ukraine; and |
| ● | other disruptions in means of transportation. |
Any significant interruption
at the combined company’s distribution facilities, an inability to transport its products to or from these facilities, or to or
from its domestic or foreign customers or suppliers, or an increase in transportation costs, for any reason, would materially adversely
affect the combined company.
Because the combined
company relies on internal and external logistics to transport its drug substances and products and medical related inventory
throughout the United States to and from its research and development center and other development hubs in U.S. locations,
it is subject to business risks and costs associated with the transportation industry. Many of these risks and costs are out of the
combined company’s control, and any of them could have a material adverse effect on its business, financial condition, and
results of operations.
The combined company relies
on a combination of internal and external logistics to transport its drug substances, drug products and medical related inventory into
and throughout the United States to and from its research and development center and other development hubs in U.S. locations.
As a result, the combined company is exposed to risks associated with the transportation industry such as weather, traffic patterns, gasoline
prices, shipping costs, foreign, and local and federal U.S. regulations, vehicular crashes, rising prices of transportation vendors,
fuel prices, taxes, license and registration fees, insurance premiums, self-insurance levels, difficulty in recruiting and retaining qualified
drivers, disruption of its technology systems, equipment supply, equipment quality, and increasing equipment and operational costs. Its
failure to successfully manage its logistics and fulfillment process could cause a disruption in its inventory supply chain and distribution,
which may adversely affect its operating results and financial condition.
The combined company relies on a limited
number of suppliers or, in some cases, single suppliers, for some of its laboratory instruments and materials and may not be able to find
replacements or immediately transition to alternative suppliers on a cost-effective basis, or at all.
The combined company sources
components of its technology from third parties and certain components are sole sourced. Obtaining substitute components may be difficult
or require the combined company to re-design its products. Any natural or other disasters, such as wild fire, earthquake, acts of war
or terrorism, shipping embargoes, labor unrest or political instability, failure in supply or other logistical channels, electrical outages
or other reasons or similar events at its third-party suppliers’ facilities that cause a loss of manufacturing capacity or a reduction
in the quality of the items manufactured would heighten the risks that the combined company faces. Changes to, failure to renew or termination
of the combined company’s existing agreements or its inability to enter into new agreements with other suppliers could result in
the loss of access to important components of the combined company’s tests and could impair, delay or suspend its commercialization
efforts. Its failure to maintain a continued and cost-effective supply of high-quality components could materially and adversely harm
its business, operating results, and financial condition.
The combined company relies on third parties
to conduct its pre-clinical and clinical studies and perform other tasks for itself. If these third parties do not successfully carry
out their contractual duties, meet expected deadlines or comply with regulatory requirements, the combined company may not be able to
obtain regulatory approval for or commercialize its product candidates and its business could be substantially harmed.
The combined company has relied
upon and plan to continue to rely upon third-party CROs to monitor and manage data for its ongoing pre-clinical and clinical programs.
The combined company relies on these parties for execution of its pre-clinical and clinical studies, and control only certain aspects
of their activities. Nevertheless, the combined company is responsible for ensuring that each of its studies is conducted in accordance
with the applicable protocol, legal, regulatory and scientific standards and its reliance on the CROs does not relieve it of its regulatory
responsibilities. The combined company and its CROs and other vendors are required to comply with current cGMP, Good Clinical Practices,
or GCP, quality system requirements, or QSR, and Good Laboratory Practices, or GLP, which are regulations and guidelines enforced by the
FDA, the Competent Authorities of the Member States of the European Economic Area, and comparable foreign regulatory authorities for all
of its product candidates in clinical development. Regulatory authorities enforce these regulations through periodic inspections of study
sponsors, principal investigators, study sites and other contractors. If the combined company or any of the combined company’s CROs
or vendors fail to comply with applicable regulations, the clinical data generated in its clinical studies may be deemed unreliable and
the FDA, EMA or comparable foreign regulatory authorities may require the combined company to perform additional clinical studies before
approving its marketing applications. It cannot assure you that upon inspection by a given regulatory authority, such regulatory authority
will determine that any of its clinical studies comply with GCP regulations. In addition, the combined company’s clinical studies
must be conducted with product candidates which are produced under cGMP regulations. The combined company’s failure to comply with
these regulations may require the combined company to repeat clinical studies, which would delay the regulatory approval process.
The combined company may not be able to
secure and maintain research institutions to conduct its clinical trials.
The combined company relies
on research institutions to conduct its clinical trials. Specifically, the limited number of centers experienced with cell therapy product
candidates heightens its dependence on such research institutions. Its reliance upon research institutions, including hospitals and clinics,
provides it with less control over the timing and cost of clinical trials and the ability to recruit subjects. If the combined company
is unable to reach agreements with suitable research institutions on acceptable terms, or if any resulting agreement is terminated, it
may be unable to quickly replace the research institution with another qualified institution on acceptable terms. The combined company
may not be able to secure and maintain suitable research institutions to conduct its clinical trials, which could have a material adverse
effect on its business and prospects.
The combined company and its collaborators
and contract manufacturers are subject to significant and ongoing regulation with respect to manufacturing its drug substances. The manufacturing
facilities on which it relies may not continue to meet regulatory requirements and have limited capacity.
Manufacturers and their facilities
are required to comply with extensive regulatory requirements, including ensuring that quality control and manufacturing procedures conform
to cGMPs. These cGMP regulations cover all aspects of manufacturing relating to its product candidates. These regulations govern manufacturing
processes and procedures (including record keeping) and the implementation and operation of quality systems to control and assure the
quality of investigational product candidates and products approved for sale. Poor control of production processes can lead to the introduction
of contaminants or to inadvertent changes in the properties or stability of its product candidates that may not be detectable in final
product testing. We, its collaborators or its contract manufacturers must supply all necessary documentation in support of an BLA or MAA
on a timely basis and must adhere to GLP and cGMP QSR regulations enforced by the FDA and other regulatory authorities through their facilities
inspection program. Some of the combined company’s contract manufacturers have never produced a commercially approved pharmaceutical
product and therefore have not obtained the requisite regulatory authority approvals to do so. The facilities and quality systems of some
or all of its collaborators and third-party contractors must pass a pre-approval inspection for compliance with the applicable regulations
as a condition of regulatory approval of its product candidates or any of its other potential product candidates. In addition, the regulatory
authorities may, at any time, audit or inspect a manufacturing facility involved with the preparation of its product candidates or its
other potential product candidates or the associated quality systems for compliance with the regulations applicable to the activities
being conducted. The combined company does not control the manufacturing process of, and are completely dependent on, its contract manufacturing
partners for compliance with the regulatory requirements. If these facilities do not pass a pre-approval plant inspection, regulatory
approval of the product candidates may not be granted or may be substantially delayed until any violations are corrected to the satisfaction
of the regulatory authority, if ever. Moreover, if the combined company’s contract manufacturer’s fail to achieve and maintain
high manufacturing standards, in accordance with applicable regulatory requirements, or there are substantial manufacturing errors, this
could result in patient injury or death, product shortages, product recalls or withdrawals, delays or failures in product testing or delivery,
cost overruns or other problems that could seriously harm its business.
Any collaboration arrangements that the
combined company may enter into in the future may not be successful, which could adversely affect its ability to develop and commercialize
its current and potential future product candidates.
The combined company may seek
collaboration arrangements with pharmaceutical or biotechnology companies for the development or commercialization of its current and
potential future product candidates. It may enter into these arrangements on a selective basis depending on the merits of retaining commercialization
rights for the combined company as compared to entering into selective collaboration arrangements with other pharmaceutical or biotechnology
companies for each product candidate, both in the United States and internationally. It will face, to the extent that the combined
company decides to enter into collaboration agreements, significant competition in seeking appropriate collaborators. Moreover, collaboration
arrangements are complex and time consuming to negotiate, document and implement. The combined company may not be successful in its efforts
to establish and implement collaborations or other alternative arrangements should the combined company so choose to enter into such arrangements.
The terms of any collaborations or other arrangements that the combined company may establish may not be favorable to itself.
Disagreements between parties
to a collaboration arrangement regarding clinical development and commercialization matters can lead to delays in the development process
or commercializing the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements
can be difficult to resolve if neither of the parties has final decision-making authority.
Collaborations with pharmaceutical
or biotechnology companies and other third parties often are terminated or allowed to expire by the other party. Any such termination
or expiration could adversely affect the combined company financially and could harm its business reputation.
The combined company’s reliance on
third parties requires it to share its trade secrets, which increases the possibility that a competitor will discover them or that its
trade secrets will be misappropriated or disclosed.
Because the combined company
relies on third parties to develop and manufacture its product candidates, the combined company must, at times, share trade secrets with
them. It seeks to protect its proprietary technology in part by entering into confidentiality agreements and, if applicable, material
transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with its collaborators, advisors,
employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights
of the third parties to use or disclose its confidential information, such as trade secrets. Despite the contractual provisions employed
when working with third parties, the need to share trade secrets and other confidential information increases the risk that such trade
secrets become known by the combined company’s competitors, are inadvertently incorporated into the technology of others, or are
disclosed or used in violation of these agreements. Given that the combined company’s proprietary position is based, in part, on
its know-how and trade secrets, a competitor’s discovery of its trade secrets or other unauthorized use or disclosure would impair
its competitive position and may have a material adverse effect on its business.
Risks Related to the Combined Company’s
Intellectual Property
The combined company has filed multiple
patent applications and have a limited number of issued patents. There can be no assurance that any of its patent applications will result
in issued patents. As a result, the combined company’s ability to protect its proprietary technology in the marketplace may be limited.
The combined company has filed
patent applications in countries worldwide. These applications cover a range of areas including: the U.S., EU, Israel and Japan. While
several patent applications have been granted, several are still pending. Unless and until its pending patent applications are issued,
their protective scope is impossible to determine. It is impossible to predict whether or how many of its patent applications will result
in issued patents. Even if pending applications are issued, they may be issued with coverage significantly narrower than what the combined
company currently seeks. For a description of the combined company’s patent applications and intellectual property, see “Item
4. Information on the Company — B. Business Overview — Intellectual Property” and “Item 4. Information
on the Company — B. Business Overview — Kadimastem Business — Intellectual Property — Methods of
Generating Glial and Neuronal Cells.”
The combined company’s proprietary
position for its product candidates currently depends upon patents protecting the method of use, which may not prevent a competitor or
other third party from using the same product candidate for another use.
The primary patent based intellectual
property protection for the combined company’s product candidates will be any patents granted on the method of use and formulation.
The combined company does not have patents or patent applications covering its products as a composition of matter (i.e., compound claims).
Composition of matter
patent claims on the active pharmaceutical ingredient, or API, in pharmaceutical products are generally considered to be the favored
form of intellectual property protection for pharmaceutical products, as such patents provide protection without regard to any
particular method of use, manufacture or formulation of the API used. Method of use patent claims protect the use of a product for
the specified method and dosing. These types of patent claims do not prevent a competitor or other third party from making and
marketing an identical API for an indication that is outside the scope of the method claims or from developing a different dosing
regimen. Moreover, even if competitors or other third parties do not actively promote their product for the combined company’s
targeted indications or uses for which it may obtain patents, physicians may recommend that patients use these products off-label,
or patients may do so themselves. Although off-label use may infringe or contribute to the infringement of method of use patents,
the practice is common and such infringement is difficult to prevent or prosecute.
There is no certainty that its pending or
future patent applications will result in the issuance of patents.
The combined company’s
success depends in part on its ability to obtain and defend patent and other intellectual property rights that are important to the commercialization
of its products and product candidates. The degree of patent protection that will be afforded to its products and processes in the U.S. and
in other important markets remains uncertain and is dependent upon the scope of protection decided upon by the patent offices, courts,
administrative bodies and lawmakers in these countries. The combined company can provide no assurance that it will successfully obtain
or preserve patent protection for the technologies incorporated into its products and processes, or that the protection obtained will
be of sufficient breadth and degree to protect its commercial interests in all countries where it conducts business. If the combined company
cannot prevent others from exploiting its inventions, it will not derive the benefit from them that it currently expects. Furthermore,
the combined company can provide no assurance that its products will not infringe patents or other intellectual property rights held by
third parties.
In Europe, for example, there
is uncertainty about the eligibility of human embryonic stem cell subject matter for patent protection. The European Patent Convention,
or EPC, prohibits the granting of European patents for inventions that concern “uses of human embryos for industrial or commercial
purposes.” A recent decision at the Court of Justice of the European Union interpreted parthenogenetically produced human embryonic
stem cells as patentable subject matter. Consequently, the European Patent Office now recognizes that human pluripotent stem cells (including
human embryonic stem cells) can be created without a destructive use of human embryos as of June 5, 2003, and patent applications
relating to human embryonic stem cell subject matter with a filing and priority date after this date are no longer automatically excluded
from patentability under Article 53 (a) EPC and Rule 28(c) EPC.
Even if the combined company will issue
patents, because the patent positions of pharmaceutical and/or biotech products are complex and uncertain, the combined company cannot
predict the scope and extent of patent protection for its product candidates.
Any patents that may be issued
to it will not ensure the protection of the combined company’s intellectual property for a number of reasons, including without
limitation the following:
| ● | any issued patents may not be broad or strong enough to prevent
competition from other drug substances including identical or similar products and technology; |
| ● | if the combined company is not issued patents or if issued
patents expire, there would be no protections against competitors making generic equivalents; |
| ● | there may be prior art of which the combined company is not
aware that may affect the validity or enforceability of a patent claim; |
| ● | there may be other patents existing, now or in the future,
in the patent landscape for PBI products, or any other product candidates that the combined company seeks to commercialize or develop,
if any, that will affect Kadimastem’s freedom to operate; |
| ● | if the combined company’s patents are challenged, a
court could determine that they are not valid or enforceable; |
| ● | a court could determine that a competitor’s technology
or product does not infringe the combined company’s patents; |
| ● | the combined company’s patents could irretrievably lapse
due to failure to pay fees or otherwise comply with regulations, or could be subject to compulsory licensing; and |
| ● | if the combined company encounters delays in its development
or clinical trials, the period of time during which it could market its products under patent protection would be reduced. |
Obtaining and maintaining the combined company’s
patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental
patent agencies, and the combined company’s patent protection could be reduced or eliminated for noncompliance with these requirements.
Periodic maintenance fees
on any issued patent are due to be paid to the U.S. PTO and foreign patent agencies in several stages over the term of the patent.
The U.S. PTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment
and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of
a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment
or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Noncompliance
events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond
to office actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In
such an event, the combined company’s competitors might be able to enter the market, which would have a material adverse effect
on its business.
The combined company may not be able to
enforce its intellectual property rights throughout the world.
Filing, prosecuting and defending
patents on product candidates in all countries throughout the world would be prohibitively expensive, and its intellectual property rights
in some countries outside the United States and Israel can be less extensive than those in the United States and Israel. In
addition, the laws of some foreign countries do not protect intellectual property to the same extent as laws in the United States
and Israel. Consequently, the combined company may not be able to seek to prevent third parties from practicing its inventions in all
countries outside the
United States and Israel,
or from selling or importing products made using its inventions in and into the United States or other jurisdictions. Competitors,
for example, may use the combined company’s technologies in jurisdictions where Kadimastem have not obtained patents to develop
their own products and further, may export otherwise infringing products to territories where it has patents, but enforcement is not as
strong as that in the United States and Israel.
Many companies have encountered
significant problems in protecting and defending intellectual property in foreign jurisdictions. The legal systems of certain countries,
particularly China and certain other developing countries, do not favor the enforcement of patents, trade secrets and other intellectual
property, particularly those relating to drug substances and product candidates and biopharmaceutical and biotechnology products, which
could make it difficult for the combined company to stop the infringement of its patents or marketing of competing products in violation
of its proprietary rights generally. To date, the combined company have not sought to enforce any issued patents in these foreign jurisdictions.
Proceedings to enforce the combined company’s patent rights in foreign jurisdictions could result in substantial costs and divert
its efforts and attention from other aspects of its business, could put its patents at risk of being invalidated or interpreted narrowly
and its patent applications at risk of not issuing and could provoke third parties to assert claims against the combined company. The
combined company may not prevail in any lawsuits that it initiates and the damages or other remedies awarded, if any, may not be commercially
meaningful. The requirements for patentability may differ in certain countries, particularly developing countries. Certain countries in
Europe and developing countries, including China and India, have compulsory licensing laws under which a patent owner may be compelled
to grant licenses to third parties. In those countries, the combined company and its licensors may have limited remedies if patents are
infringed or if it or its licensors are compelled to grant a license to a third party, which could materially diminish the value of those
patents. This could limit its potential revenue opportunities. Accordingly, its efforts to enforce its intellectual property rights around
the world may be inadequate to obtain a significant commercial advantage from the intellectual property that it develops or licenses.
If the combined company is unable to maintain
effective proprietary rights for its product candidates, it may not be able to compete effectively in its markets.
In addition to the
protection afforded by any patents currently owned and that may be granted, historically, it has relied on trade secret protection
and confidentiality agreements to protect proprietary know-how that is not patentable or that the combined company elects not to
patent, processes that are not easily known, knowable or easily ascertainable, and for which patent infringement is difficult to
monitor and enforce and any other elements of its product candidate discovery and development processes that involve proprietary
know-how, information or technology that is not covered by patents. However, trade secrets can be difficult to protect. The combined
company seeks to protect its proprietary technology and processes, in part, by entering into confidentiality agreements with its
employees, consultants, scientific advisors, and contractors. It also seeks to preserve the integrity and confidentiality of its
data, trade secrets and intellectual property by maintaining physical security of its premises and physical and electronic security
of its information technology systems. Agreements or security measures may be breached, and the combined company may not have
adequate remedies for any breach. In addition, the combined company’s trade secrets and intellectual property may otherwise
become known or be independently discovered by competitors.
The combined company cannot
provide any assurances that its trade secrets and other confidential proprietary information will not be disclosed in violation of its
confidentiality agreements or that competitors will not otherwise gain access to its trade secrets or independently develop substantially
equivalent information and techniques. Also, misappropriation or unauthorized and unavoidable disclosure of its trade secrets and intellectual
property could impair its competitive position and may have a material adverse effect on its business. Additionally, if the steps taken
to maintain its trade secrets and intellectual property are deemed inadequate, it may have insufficient recourse against third parties
for misappropriating any trade secret.
Third parties may initiate legal proceedings
alleging that the combined company is infringing their intellectual property rights, the outcome of which would be uncertain and could
have a material adverse effect on the success of its business.
The combined company’s
commercial success depends upon its ability to develop, manufacture, market and sell its platform technology without infringing the proprietary
rights of third parties. There is considerable intellectual property litigation in the drug substances and product candidates and pharmaceutical
industries. While no such litigation has been brought against it and the combined company has not been held by any court to have infringed
a third party’s intellectual property rights, the combined company cannot guarantee that its technology or use of its technology
does not infringe third-party patents. It is also possible that it has failed to identify relevant third-party patents or applications.
For example, applications filed before November 29, 2000, and certain applications filed after that date that will not be filed outside
the United States remain confidential until patents issue. Patent applications in the United States and elsewhere are published
approximately 18 months after the earliest filing, which is referred to as the priority date. Therefore, patent applications covering
its technology could have been filed by others without its knowledge. Additionally, pending patent applications which have been published
can, subject to certain limitations, be later amended in a manner that could cover the combined company’s technology.
The combined company may become party to,
or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to its technology,
including inter parties review, interference, or derivation proceedings before the U.S. PTO and similar bodies in other countries.
Third parties may assert infringement claims against the combined company based on existing intellectual property rights and intellectual
property rights that may be granted in the future.
If the combined company is
found to infringe a third party’s intellectual property rights, the combined company could be required to obtain a license from
such third party to continue developing and marketing its technology. However, the combined company may not be able to obtain any required
license on commercially reasonable terms or at all. Any inability to secure licenses or alternative technology could result in delays
in the introduction of its products or lead to prohibition of the manufacture or sale of products by it. Even if the combined company
were able to obtain a license, the combined company could be non-exclusive, thereby giving its competitors access to the same technologies
licensed to it. The combined company could be forced, including by court order, to cease commercializing the infringing technology. In
addition, the combined company could be found liable for monetary damages, including treble damages and attorneys’ fees if the combined
company is found to have willfully infringed a patent. A finding of infringement could prevent the combined company from commercializing
its technology or force the combined company to cease some of its business operations, which could materially harm the combined company’s
business. Claims that the combined company has misappropriated the confidential information or trade secrets of third parties could have
a similar negative impact on its business.
The combined company may be subject to claims
challenging the inventorship of its patents and other intellectual property.
The combined company may be
subject to claims that former employees, collaborators or other third parties have an interest in its patents or other intellectual property
as an inventor or co-inventor. For example, the combined company may have inventorship disputes arise from conflicting obligations of
consultants or others who are involved in developing its product candidates. Litigation may be necessary to defend against these and other
claims challenging inventorship. If the combined company fails in defending any such claims, in addition to paying monetary damages, it
may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such
an outcome could have a material adverse effect on its business. Even if it is successful in defending against such claims, litigation
could result in substantial costs and be a distraction to management and other employees. In addition, the combined company may receive
less revenue from future products if any of its employees successfully claim for compensation for their work in developing the combined
company’s intellectual property, which in turn could impact the combined company’s future profitability.
Under applicable employment laws, the combined
company may not be able to enforce covenants not to compete and therefore may be unable to prevent its competitors from benefiting from
the expertise of some of its former employees. In addition, employees may be entitled to seek compensation for their inventions irrespective
of their agreements with it.
The combined company generally
enters into non-competition agreements with its employees and certain key consultants. These agreements prohibit employees and certain
key consultants, if they cease working for the combined company, from competing directly with it or working for its competitors or clients
for a limited period of time. The combined company may be unable to enforce these agreements under the laws of the jurisdictions in which
the combined company’s employees work and it may be difficult for it to restrict its competitors from benefitting from the expertise
its former employees or consultants developed while working for it. For example, Israeli courts have required employers seeking to enforce
non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm one of a
limited number of material interests of the employer which have been recognized by the courts, such as the secrecy of a company’s
confidential commercial information or the protection of its intellectual property. If the combined company cannot demonstrate that such
interests will be harmed, it may be unable to prevent its competitors from benefiting from the expertise of its former employees or consultants
and its ability to remain competitive may be diminished.
In addition, under Israeli
law, if the combined company wishes to obtain ownership over inventions developed by its employees, which inventions were developed while
performing their employment activities, but outside the performance of their contractual duties, the combined company is required to compensate
the employee for the rights to their respective inventions. There can be no guarantee that the combined company will be able to obtain
any such inventions and the failure to obtain such ownership rights over employee inventions could have a material adverse effect on its
operations and ability to effectively compete.
Risks Related to Israeli Law and its Operations
in Israel
Potential political, economic and military
instability in the State of Israel, where its headquarters, members of its management team, its production and research and development
facilities are located, may adversely affect its results of operations.
The combined company’s
executive offices, research and development laboratories and manufacturing facility are located in Ness-Ziona, Israel. In addition, the
majority of its key employees, officers and directors are residents of Israel. Accordingly, political, economic and military conditions
in Israel may directly affect its business. Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken
place between Israel and groups in its neighboring countries, including Hamas (an Islamist militia and political group that has historically
controlled the Gaza Strip) and Hezbollah (an Islamist militia and political group based in Lebanon). In addition, several countries, principally
in the Middle East, restrict doing business with Israel, and additional countries may impose restrictions on doing business with Israel
and Israeli companies whether as a result of hostilities in the region or otherwise. Any hostilities involving Israel, terrorist activities,
political instability or violence in the region or the interruption or curtailment of trade or transport between Israel and its trading
partners could adversely affect its operations and results of operations and the market price of its Common Shares.
Conditions in the Middle East and in Israel,
where its research and development facilities are located, may harm its operations.
The combined company’s
office where the combined company conducts its research and development, operations, sales outside the Americas, and administration activities,
is located in Israel. Many of the combined company’s employees are residents of Israel. Most of the combined company’s executive
officers and directors are residents of Israel. Accordingly, political, economic and military and security conditions in Israel and the
surrounding region may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed conflicts
have taken place between Israel and its neighboring countries, and between Israel and the Hamas (an Islamist militia and political group
in the Gaza Strip) and Hezbollah (an Islamist militia and political group in Lebanon). Ongoing and revived hostilities in the Middle East
or other Israeli political or economic factors, could harm our operations.
In particular, in October 2023,
Hamas terrorists infiltrated Israel’s southern border from the Gaza Strip and conducted a series of attacks on civilian and military
targets. Hamas also launched extensive rocket attacks on the Israeli population and industrial centers located along Israel’s border
with the Gaza Strip and in other areas within the State of Israel. Moreover, both Hamas and Hezbollah are designated as terrorist organizations
by the United States, the European Union, Canada, and Israel, reinforcing the international consensus on their use of terror tactics and
destabilizing activities. These attacks resulted in thousands of deaths and injuries, and Hamas additionally kidnapped many Israeli civilians
and soldiers. Following the attack, Israel’s security cabinet declared war against Hamas and commenced a military campaign against
Hamas and Hezbollah and these terrorist organizations in parallel continued rocket and terror attacks. As of May 15, 2025, the ceasefire
with Hamas that had been in place since January 2025 has ended and hostilities have resumed. Other regional hostilities, since October
7, 2023, concurrently became more pronounced. This includes and has included a northern front war between Israel and Hezbollah, which
as of May 15, 2025 is subject to a ceasefire between Israel and Lebanon, hostilities between Israel and Iran, and a continued conflict
between Israel and the Houthi Movement in Yemen, which have included strikes against Israel and subsequent retaliation by Israel. Iran
is also believed to be developing nuclear weapons and to have a strong influence among extremist groups in the region, such as Hamas in
Gaza, Hezbollah in Lebanon, the Houthi movement in Yemen and various rebel militia groups in Syria. These situations may potentially escalate
in the future to more violent events which may affect Israel and the combined company.
In connection with the current
multi-front conflict, Israeli military reservists have been called up to perform military service. As of the date of this annual report,
the combined company has not been impacted by any absences of personnel at its service providers or counterparties located in Israel.
Military service call ups that result in absences of personnel from the combined company for an extended period of time may materially
and adversely affect its business, prospects, financial condition and results of operations.
Since the war broke out on
October 7, 2023, the combined company’s operations have not been adversely affected by this situation, and it has not experienced
any material disruptions to its operations. The combined company has the ability, if necessary, to shift its manufacturing from Israel
to other countries where it has business partners, and it have not had customers in Israel in the last year. However, the intensity and
duration of the war in the Middle East is difficult to predict at this stage, as are such war’s economic implications on the combined
company’s business and operations and on Israel’s economy in general. if the war in the other fronts, such as Lebanon, Syria
and the West Bank expands further, the combined company’s operations may be adversely affected.
Any armed conflicts,
terrorist activities or political instability in the region could adversely affect business conditions, could harm the combined
company’s results of operations and could make it more difficult for the combined company to raise capital. Parties with whom
the combined company does business may decline to travel to Israel during periods of heightened unrest or tension, forcing the
combined company to make alternative arrangements when necessary in order to meet the combined company’s business partners
face to face. In addition, the political and security situation in Israel may result in parties with whom the combined company has
agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements
pursuant to force majeure provisions in such agreements. Further, in the past, the State of Israel and Israeli companies have been
subjected to economic boycotts. Several countries still restrict business with the State of Israel and with Israeli companies. These
restrictive laws and policies may have an adverse impact on the combined company’s operating results, financial condition or
the expansion of its business.
Any hostilities involving
Israel or the interruption or curtailment of trade between Israel and its trading partners could adversely affect the combined company’s
operations and results of operations. In recent years, the hostilities involved missile strikes against civilian targets in various
parts of Israel, including areas in which the combined company’s employees and some of its consultants are located, and negatively
affected business conditions in Israel.
The combined company’s
commercial insurance does not cover losses that may occur as a result of events associated with the security situation in the Middle East.
Although the Israeli government currently covers the reinstatement value of direct damages that are caused by terrorist attacks or acts
of war, the combined company cannot assure you that this government coverage will be maintained. Any losses or damages incurred by the
combined company could have a material adverse effect on its business. Any armed conflicts or political instability in the region would
likely negatively affect business conditions and could harm the combined company’s results of operations.
Although the Israeli government
currently covers the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, we cannot assure you that
this government coverage will be maintained or that it will sufficiently cover the combined company’s potential damages. Any losses
or damages incurred by the combined company could have a material adverse effect on the combined company’s business. Any armed conflicts
or political instability in the region would likely negatively affect business conditions and could harm the combined company’s
results of operations.
Several countries, principally
in the Middle East, still restrict doing business with Israel and Israeli companies, and additional countries may impose restrictions
on doing business with Israel and Israeli companies, whether as a result of hostilities in the region or otherwise. In addition, there
have been increased efforts by certain countries, activists and organizations to cause companies, research institutions and consumers
to boycott Israeli goods and cooperation with Israeli-related entities based on Israeli government policies. Such actions, particularly
if they become more widespread, may adversely impact the combined company’s ability to collaborate with other third parties.
Additionally, political conditions
within Israel may affect the combined company’s operations. Many citizens and groups have responded strongly against the Israeli
government’s pursuit of judicial reform. To date, these initiatives have been substantially put on hold. Actual or perceived instability
in Israel or any negative changes in the political environment may individually or in the aggregate adversely affect the combined company.
Finally, political conditions
within Israel may affect the combined company’s operations. Israel has held five general elections between 2019 and 2022, and prior
to October 2023, the Israeli government pursued extensive changes to Israel’s judicial system, which sparked extensive political
debate and unrest. To date, these initiatives have been substantially put on hold. Actual or perceived political instability in Israel
or any negative changes in the political environment, may individually or in the aggregate adversely affect the Israeli economy and, in
turn, the combined company’s business, financial condition, results of operations and growth prospects.
Exchange rate fluctuations between the U.S. Dollar
and New Israeli Shekels, or NIS, may negatively affect the combined company’s earnings and could adversely affect its results of
operations.
The combined company incurs
expenses in NIS, U.S. dollars, and Euros, but the combined company’s financial statements are denominated in NIS as its financial
currency. Accordingly, the combined company faces exposure to adverse movements in currency exchange rates. Accordingly, the combined
company faces exposure to adverse movements in the currency exchange rate of the NIS against these foreign currencies, which may have
a negative effect on its revenue and costs. If the NIS weakens against any of these currencies, the translation of these foreign currency
denominated transactions will result in decreased revenue in NIS. Changes in currency exchange rates may have a negative effect on
its financial results.
The termination or reduction of tax and
other incentives that the Israeli government provides to Israeli companies may increase the combined company’s costs and taxes.
The Israeli government currently
provides tax and capital investment incentives to Israeli companies, as well as grant and loan programs relating to research and development
and marketing and export activities. In recent years, the Israeli government has reduced the benefits available under these programs
and the Israeli governmental authorities may in the future further reduce or eliminate the benefits of these programs. The combined company
may take advantage of these benefits and programs in the future; however, there can be no assurance that such benefits and programs will
be available to it. If it qualifies for such benefits and programs and fail to meet the conditions thereof, the benefits could be cancelled
and it could be required to refund any benefits it might already have enjoyed and become subject to penalties. Additionally, if the combined
company qualifies for such benefits and programs and they are subsequently terminated or reduced, it could have an adverse effect on the
combined company’s financial condition and results of operations.
The combined company may be required to
pay monetary remuneration to its Israeli employees for their inventions, even if the rights to such inventions have been duly assigned
to us.
The combined company enters
into agreements with its Israeli employees pursuant to which such individuals agree that any inventions created in the scope of their
employment are either owned exclusively by the combined company or are assigned to it, depending on the jurisdiction, without the employee
retaining any rights. A portion of the combined company’s intellectual property has been developed by its Israeli employees during
their employment for it. Under the Israeli Patent Law, 5727-1967, or the Patent Law, inventions conceived by an employee during the
course of his or her employment and within the scope of said employment are considered “service inventions. Service inventions belong
to the employer by default, absent a specific agreement between the employee and employer otherwise. The Patent Law also provides that
if there is no agreement regarding the remuneration for the service inventions, even if the ownership rights were assigned to the employer,
the Israeli Compensation and Royalties Committee, or the Committee, a body constituted under the Patent Law, shall determine whether the
employee is entitled to remuneration for these inventions. The Committee has not yet determined the method for calculating this Committee-enforced
remuneration. While it has previously been held that an employee may waive his or her rights to remuneration in writing, orally or by
conduct, litigation is pending in the Israeli labor court is questioning whether such waiver under an employment agreement is enforceable.
Although the combined company’s Israeli employees have agreed that the combined company exclusively own any rights related to their
inventions, the combined company may face claims demanding remuneration in consideration for employees’ service inventions. As a
result, the combined company could be required to pay additional remuneration or royalties to its current and/or former employees, or
be forced to litigate such claims, which could negatively affect the combined company’s business.
The combined company received Israeli government
grants for certain of its research and development activities, the terms of which may require it to pay royalties and to satisfy specified
conditions in order to manufacture products and transfer technologies outside of Israel. If the combined company fails to satisfy these
conditions, it may be required to pay penalties and refund grants previously received.
The combined company’s
research and development efforts have been financed in part through royalty-bearing grants that it received from the Israel Innovation
Authority, or the IIA. The combined company is further required to comply with the requirements of the Israeli Encouragement of Industrial
Research, Development and Technological Innovation Law, 5744-1984, as amended, and related regulations, or the Research Law, with
respect to those past grants. When a company develops know-how, technology or products using IIA grants, the terms of these grants and
the Research Law restrict the transfer or license of such know-how, and the transfer of manufacturing or manufacturing rights of such
products, technologies or know-how outside of Israel, without the prior approval of the IIA. Therefore, the discretionary approval
of an IIA committee would be required for any transfer or license to third parties inside or outside of Israel of know how or for the
transfer outside of Israel of manufacturing or manufacturing rights related to those aspects of such technologies. The combined company
may not receive those approvals. Furthermore, the IIA may impose certain conditions on any arrangement under which it permits the combined
company to transfer technology or development.
The transfer or license
of IIA-supported technology or know-how outside of Israel and the transfer of manufacturing of IIA-supported products, technology or
know-how outside of Israel may involve the payment of significant amounts, depending upon the value of the transferred or licensed
technology or know-how, the combined company’s research and development expenses, the amount of IIA support, the time of
completion of the IIA-supported research project and availability of similar services in Israel and other factors. These
restrictions and requirements for payment may impair its ability to sell, license or otherwise transfer its technology assets
outside of Israel or to outsource or transfer development or manufacturing activities with respect to any product or technology
outside of Israel. Furthermore, the consideration available to the combined company’s shareholders in a transaction involving
the transfer outside of Israel of technology or know-how developed with IIA funding (such as a merger or similar transaction) may be
reduced by any amounts that the combined company are required to pay to the IIA.
The combined company may not be able to
enforce covenants not-to-compete under current Israeli law that might result in added competition for its products.
The combined company have
non-competition agreements with all of its employees, all of which are governed by Israeli law. These agreements prohibit the combined
company employees from competing with or working for its competitors. However, Israeli courts are reluctant to enforce non-compete undertakings
of former employees and tend, if at all, to enforce those provisions for relatively brief periods of time in restricted geographical areas,
and only when the employee has obtained unique value to the employer specific to that employer’s business and not just regarding
the professional development of the employee. If it is not able to enforce non-compete covenants, it may be faced with added competition.
ITEM 4. INFORMATION ON THE COMPANY
A. History and Development of the Company
We were established on June
10, 2015 as a Swiss limited company. Our registered office is located at The Circle 6, Postfach, 8058 Zurich, Switzerland.
In 2016, we acquired several
patents from Assistance Publique - Hopitaux de Paris, or AP-HP, in France, including for our lead compound mazindol. Since our founding,
we have assembled an experienced leadership team with a track record of developing and repurposing products to treat rare neurological
disorders. Our Chief Executive Officer, Alexander Zwyer, has held a variety of senior leadership roles of increasing responsibility in
the pharmaceutical industry, including global roles in business development, sales and marketing, medical as well as regulatory affairs,
and general management. We complement our management team with a group of scientific and clinical advisors that includes internationally
recognized North American and European experts in CNS disorders, including the areas of our current focus, rare hypersomnolence (specifically,
narcolepsy) and complex neurodevelopmental disorders.
Our principal executive offices
are located The Circle 6, Postfach, 8058 Zurich, Switzerland, our general telephone number is (+41) 44 512 21 50 and our internet address
is www.nlspharma.com. The SEC also maintains an internet website that contains reports, proxy and information statements, and other information
regarding issuers that file electronically with the SEC. Our filings with the SEC are available to the public through the SEC’s
website at www.sec.gov. The information contained on our website and the SEC website or available through such websites is not incorporated
by reference into and should not be considered a part of this annual report, and the references to websites in this annual report are
inactive textual references only.
We are an EGC. As such, we
are eligible to, and intend to, take advantage of certain exemptions from various reporting requirements applicable to other public companies
that are not “emerging growth companies” such as not being required to comply with the auditor attestation requirements of
Section 404 of the Sarbanes-Oxley Act. We could remain an EGC for up to five years, or until the earliest of (a) the last day of the first
fiscal year in which our annual gross revenues exceeds $1.235 billion, (b) the date that we become a “large accelerated filer”
as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our common shares that is held by non-affiliates
exceeds $700 million as of the last business day of our most recently completed second fiscal quarter, or (c) the date on which we have
issued more than $1 billion in nonconvertible debt during the preceding three-year period.
We are a foreign private
issuer as defined by the rules under the Securities Act and the Exchange Act. Our status as a foreign private issuer also exempts us
from compliance with certain laws and regulations of the SEC and certain regulations of Nasdaq, including the proxy rules, the
short-swing profits recapture rules, and certain governance requirements such as independent director oversight of the nomination of
directors and executive compensation. In addition, we are not required to file annual, quarterly and current reports and financial
statements with the SEC as frequently or as promptly as U.S. domestic companies registered under the Exchange Act.
Starting the last quarter
of 2023, we began to adjust our workforce, primarily comprised of consultants. On March 20, 2024, we announced that we had secured Exclusive
Global License for Next-Generation Non-Sulfonamide Dual Orexin Agonist Platform. On March 22, 2024, we announced the closing of $1.75
Million Registered Direct Offering.
Agreement and Plan of Merger
with Kadimastem
On November 4, 2024, the Company,
Merger Sub and Kadimastem entered into the Merger Agreement, pursuant to which (i) Merger Sub will merge with and into Kadimastem, with
Kadimastem as the surviving company, and (ii) at the effective time of the Merger, or the Effective Time, each issued and outstanding
Kadimastem Ordinary Share, will be exchanged for and automatically converted into the right to receive from the Company that certain number
of fully paid and nonassessable common shares as calculated in accordance with the terms of the Merger Agreement, or the Exchange Ratio.
The initial Exchange Ratio fully diluted share split resulted in Kadimastem shareholders holding approximately 85% of the issued and outstanding
common shares, subject to certain adjustments as of the closing of the Merger, or the Closing, including as a result of estimated closing
cash of NLS and Kadimastem and estimated closing indebtedness of NLS. The target fully diluted share split of 85% / 15% is subject to
adjustment pursuant to the terms of the Merger Agreement, including as a result of estimated closing cash of NLS and Kadimastem and estimated
closing indebtedness of NLS.
The Merger Agreement provides
that, upon the terms and subject to the conditions thereof, following the Closing, the Company shall work diligently to dispose of any
intellectual property, assets, rights, contracts, agreements, leases, arrangements (regardless of form), approvals, licenses, permits,
whether current or future, whether or not contingent, of the Company and its subsidiaries related solely to any product candidate of the
Company and its subsidiaries including Quilience and Nolazol, but other than the Company’s Dual Orexin Agonist platform, or such
assets to be disposed, the Legacy Assets. It is expected that the proceeds from any such disposition will be distributed to the shareholders
and warrantholders of the Company as of immediately prior to the Effective Time pursuant to the terms and conditions of the CVR Agreement,
subject to the adjustments set forth therein.
At the Effective Time, each:
| ● | Kadimastem
Ordinary Share issued and outstanding immediately prior to the Effective Time will be exchanged for and converted into the right to receive
a number of newly issued, fully paid and nonassessable Common Shares equal to the Exchange Ratio; |
| ● | option,
restricted share unit, restricted share, warrant or other rights issued and outstanding, whether vested or unvested, to purchase Kadimastem
Ordinary Shares, shall be assumed by the Company and converted into an option, warrant, other award, or right, as applicable, to purchase
Common Shares in accordance with the terms of the Merger Agreement; and |
| ● | each
Common Share issued and outstanding immediately prior to the Effective time, and each Common Share acquirable upon the exercise of outstanding
warrants and pre-funded warrants of the Company, shall continue to remain outstanding and, in addition, be entitled to a contingent value
right, or CVR, pursuant to the terms of the Merger Agreement and the CVR Agreement. |
In addition, several convertible
loans issued to Kadimastem will be converted into equity securities of Kadimastem upon Closing, including (1) a loan to the Company from
certain lenders, including Alpha Capital Anstalt, in the amount of $1.7 million; and (2) a loan from Michel Revel in the amount of
NIS 3.4 million. Under the Merger Agreement, any shareholder receiving common shares in excess of a 9.99% beneficial ownership limitation
as a result of the Merger, shall be issued instead pre-funded warrants exercisable for a number of common shares in excess of the
beneficial ownership limitation, at an exercise price equal to the par value of the common shares as of the Effective Time, which, in
any event, shall be no less than CHF 0.0001 per share.
The Merger Agreement and the
consummation of the transactions contemplated thereby have been approved by the Company’s board of directors, or the Board, and
Kadimastem’s board of directors and shareholders. The Board has resolved, subject to customary exceptions, to recommend that the
shareholders of the Company approve the Merger Agreement and the transactions contemplated therein.
The Merger Agreement contains
representations, warranties and covenants by the Company, Merger Sub, and Kadimastem that are customary for a transaction of this nature,
including among others, covenants regarding the conduct of the Company’s business during the pendency of the transactions contemplated
by the Merger Agreement, the conduct of Kadimastem’s business during the pendency of the transactions contemplated by the Merger
Agreement, public disclosures and the use of reasonable best efforts to make all filings required by governmental entities following the
execution of the Merger Agreement. In addition, certain covenants require each of the parties to use, subject to the terms and conditions
of the Merger Agreement, their reasonable best efforts to consummate the Merger.
The Merger Agreement contains
closing conditions that are customary for a transaction of this nature, including the requirement for approval by the shareholders of
each of Kadimastem and the Company. The Merger has been approved by the shareholders of Kadimastem and is expected to be voted upon by
the shareholders of the Company. In addition, the Merger Agreement requires the Company to have paid off, redeemed or satisfied all of
its trade and vendor payables, and all of its debts to its officers, directors and shareholders. The Merger Agreement requires the Company
to have at least $600,000 in cash at the Closing and requires Kadimastem to have at least $3,500,000 in cash at the Closing, in each case
subject to adjustments as set forth in the Merger Agreement.
The Exchange Ratio will be determined based on
a formula that is expected to result in the number of issued and outstanding Kadimastem Ordinary Shares (calculated on a fully-diluted
basis, inclusive of Kadimastem Ordinary Shares resulting from the conversion of Kadimastem’s equity awards) being exchanged for
that certain number of newly issued Common Shares that will equal 85% of all issued and outstanding shares (calculated on a fully-diluted
basis, inclusive of Kadimastem’s equity awards assumed by NLS), subject to the adjustments as set forth in the Merger Agreement,
including: (i) the amount of any proceeds received by NLS in connection with the sale of Common Shares to investors introduced to
NLS by Kadimastem or its representatives, in each case during the period following the Signing Date (as defined in the Merger Agreement)
up to and including the Closing (as defined in the Merger Agreement), or the Investment Proceeds Adjustment, (ii) the amount by which
NLS’s estimate of its cash at the Closing differs from the target of $600,000, subject to the Investment Proceeds Adjustment, (iii) the
amount by which NLS’s estimate of its indebtedness at the Closing differs from the target of $0, and (iv) the amount by which
Kadimastem’s estimate of its cash at the Closing differs from the target of $3,500,000, subject to the Investment Proceeds Adjustment.
Based on the cash proceeds from the Company’s recent financing transactions dated October 2024 through March 2025 (approximately
$5.7 million mostly from investors introduced to NLS by Kadimastem, which satisfy the Investment Proceeds Adjustment) (see “Item
5. Operating and Financial Review and Prospects — Components of Operating Results — Recent Financing Agreements”)
the parties currently estimate the fully diluted share split at the Closing will be 80% to Kadimastem shareholders and 20% to NLS shareholders.
The Merger Agreement includes
covenants requiring the Company and its representatives not to (i) initiate, seek, solicit or knowingly encourage (including by way of
furnishing any non-public information relating to the Company or any of its subsidiaries), or knowingly induce or take any other action
which would reasonably be expected to lead to the making, submission or announcement of any Parent Acquisition Proposal (as defined in
the Merger Agreement), (ii) engage in negotiations or discussions with, or provide any non-public information or non-public data to, any
person (other than Kadimastem or any of its affiliates or any of its representatives) relating to any Parent Acquisition Proposal or grant
any waiver or release under any standstill or other agreement (except that if the Board (or any committee thereof) determines in good
faith that the failure to grant any waiver or release would be inconsistent with the Company’s directors’ fiduciary duties
under applicable law, the Company may waive any such standstill provision in order to permit a third party to make a Parent Acquisition
Proposal), (iii) enter into any agreement, including any letter agreement, memorandum of understanding, agreement in principle merger
agreement, or similar agreement relating to any Parent Acquisition Proposal, or (iv) otherwise resolve to do any of the foregoing.
Notwithstanding these
restrictions, the Company may under certain circumstances provide information to and engage in discussions or negotiations with
third parties with respect to an acquisition proposal that the Board determines in good faith, after consultation with its financial
advisor and outside legal counsel, constitutes or is reasonably likely to constitute or lead to a Parent Superior Proposal (as
defined in the Merger Agreement). In addition, the Board is permitted, subject to the terms and conditions set forth in the Merger
Agreement, to make a Parent Adverse Recommendation Change (as defined in the Merger Agreement).
The Merger Agreement contains
customary termination rights for each of the Company and Kadimastem. On January 30, 2025, the Merger Agreement was amended such that the
amended Merger Agreement includes the right of the Company and Kadimastem to terminate the Merger Agreement if the Closing shall not have
occurred on or before April 30, 2025, which outside date can be further extended by mutual agreement. On May 5, 2025, the Merger Agreement
was further amended to further extend the outside date to June 30, 2025. The Merger Agreement also provides that the Company shall pay
to Kadimastem a termination fee of $10,000,000 plus the Company Operating Expenses (as defined in the Merger Agreement), up to a maximum
of $250,000 per month beginning July 28, 2024, and the Transaction Expenses (as defined in the Merger Agreement) if the Company terminates
the Merger Agreement prior to obtaining the Parent Requisite Vote (as defined in the Merger Agreement) to enter into a definitive agreement
providing for a Parent Superior Proposal (as defined in the Merger Agreement) in accordance with terms of the Merger Agreement.
For a more comprehensive discussion
of the risks related to the Merger, please see under the section “Risk Factors - Risks Related to the Merger.”
Contingent Value Right
Agreement
Prior to the Closing, the
Company will enter into the CVR Agreement with VStock Transfer, LLC, which will govern the terms of the CVRs. Each CVR will represent
the right to additional payments based on the proceeds, subject to certain adjustments, received by the Company from the disposition of
the Legacy Assets.
The right to the CVRs as evidenced
by the CVR Agreement is a contractual right only and will not be transferable, except in the limited circumstances specified in the CVR
Agreement.
Support Agreement
Concurrently with the execution
of the Merger Agreement, the Company entered into support agreements, each, a Support Agreement with certain shareholders and of the Company,
or the Supporting Persons, covering approximately 40% of the outstanding Common Shares as of January 31, 2025. Pursuant to the Support
Agreements, each Supporting Person has agreed, among other things, to vote its Common Shares, and any other voting securities such Supporting
Person might hold, in favor of (A) (i) the issuance of Common Shares equal to the required number of Common Shares to serve as the
Merger Consideration, and (ii) an ordinary capital increase under Swiss law, excluding the subscription rights of the existing holders
of Common Shares, for the purpose of making available the required number of Common Shares to serve as the Merger Consideration, and (B)
any proposal to adjourn or postpone the shareholder meeting called to vote upon resolutions (A)(i) and (ii) to a later date if there are
not sufficient votes to approve resolutions (A)(i) and (ii). The Support Agreement will terminate upon the earliest to occur of (a) the
date that the Shareholder Resolutions (as defined in the Support Agreements) are approved, (b) the termination of the Merger Agreement
in accordance with its terms, (c) written notice of termination of the Support Agreement by the Company to the Supporting Person, and
(d) 365 days from the Effective Date (as defined in the Support Agreements).
In 2024, 2023 and 2022
our capital expenditures amounted to $0, $0, and $0, respectively. Our current capital expenditures are primarily for software and office
furniture substantially all in Switzerland, and we expect to finance these expenditures primarily from cash on hand.
B. Business Overview
We are a clinical-stage
biopharmaceutical company focused on the discovery and development of innovative therapies for patients with rare and complex CNS
disorders with unmet medical needs. Our lead compound mazindol, a triple monoamine reuptake inhibitor and partial orexin receptor 2
agonist, in a proprietary ER formulation, is being developed for the treatment of narcolepsy (lead indication), IH (follow-on
indication) and potentially ADHD (back-up indication). We believe that this unique mechanism of action will also enable Mazindol ER
to provide potential therapeutic benefit in other rare and complex CNS disorders. CNS disorders are a diverse group of conditions
that include neurological, psychiatric, and substance use disorders. According to the World Health Organization and based on data
from the Global Burden of Disease Report, CNS disorders result in a socio-economic burden of more than $317 billion annually in the
United States alone. Additionally, CNS disorders were expected to account for approximately 15% of the global disease burden in
2020, the largest of any disease area. However, treatment options for these conditions are often limited, inadequate or
non-existent, and the development of new CNS treatments generally trails behind other therapeutic areas. We are pursuing the
development of the next generation of CNS therapies with high medical impact to address this critical and growing unmet need. Our
dual development strategy is designed to optimize the outcome of our clinical programs by developing new chemical entities, or
NCE’s, from known molecules with strong scientific rationale, and also by re-defining previously approved molecules with
well-established tolerability and safety profiles, as determined by applicable regulatory agencies. We believe that our streamlined
clinical development approach has the potential to advance our product candidates rapidly through early-stage clinical trials, while
carrying an overall lower development risk. A lower development risk, we believe, exists with respect to the development of our lead
product candidate, Quilience, and follow-on product candidate, due to their use of mazindol as the active ingredient, which was
previously approved and marketed in the United States, Japan and Europe to manage exogenous obesity (obesity caused by excessive
eating).
Our discovery platform currently
focuses on single molecules that operate through multiple mechanisms designed to target the complexity of the CNS disease state, and,
we believe these may potentially offer new treatment options for patients, including for those patients who are refractory to currently
available treatments. We recently announced pre-clinical results of NLS-4, our next-generation wake-promoting drug candidate, for the
chronic fatigue syndrome, or CFS, associated with the symptoms of Long-COVID, also known as Chronic Fatigue caused by COVID -19 infection.
Our current focus is in the
therapeutic areas of rare hypersomnia disorders (conditions highlighted by EDS) and complex neurodevelopmental disorders, and includes
our lead product candidate: Quilience, for the treatment of EDS and cataplexy associated with narcolepsy, and our follow-on candidate
Nolazol, for the treatment of ADHD. We initiated our clinical development with a Phase 2 clinical trial in the third quarter of 2021,
in adult patients with narcolepsy. We published positive interim top-line results from our Phase 2 clinical trial in March 2022. As a
result, we intend to apply for expedited development program(s) facilitated by the FDA, such as Breakthrough Therapy and/or Fast Track
designations and by the European Medicines Agency, or EMA, such as PRIME. We have completed a Phase 2 clinical trial evaluating the safety
and efficacy of Nolazol in adults with ADHD in the U.S. Although further clinical development of Nolazol in ADHD is on hold, given the
positive outcome of this trial, we may initiate Phase 3 clinical trials after we receive approval to commercialize Quilience. We also
intend to seek FDA and other regulatory approval for Nolazol for use in children with ADHD, which requires additional nonclinical work,
as well as staged clinical work in determining safe dosing and monitoring. In addition, following our current focus on the development
of Quilience for narcolepsy in adults, and if approved for marketing, we intend to seek a label expansion for the treatment of narcolepsy
in pediatric patients, which may require additional pre-clinical and clinical studies.
Quilience and Nolazol both
contain mazindol as the active ingredient in a proprietary controlled release, or CR, formulation developed for a once-a-day dosing. Mazindol
has a well-established safety record from its extended history of clinical use across the United States and several countries in Europe,
where mazindol was previously approved in an immediate release formulation for the short-term management of exogenous obesity. It was
marketed for nearly 30 years, into the early 2000’s, before being voluntarily withdrawn from the market not for safety nor for efficacy
reasons (Docket n° FDA-2007-P-0326) but for commercial. In addition to the 30-year period in which it was marketed, mazindol was also
widely used off-label and prescribed under compassionate use for the treatment of narcolepsy for approximately four decades, during which
time it demonstrated a well-tolerated safety profile in patients over long-term, chronic use of the drug. We have entered into an agreement
with Novartis Pharma AG for the exclusive rights to mazindol pre-clinical, non-clinical and clinical data and to Sanorex (mazindol) NDA
in the U.S., and non-exclusive rights of mazindol in the rest of the world, except Japan and intend to use the toxicology, clinical safety
and tolerability, and CMC intellectual property from the Sanorex (mazindol) NDA to support a marketing application for Mazindol ER for
the treatment of narcolepsy.
We believe that our lead product
candidate, Quilience, offers a meaningfully differentiated product profile over current treatment options for the following reasons:
| ● | Mechanism
of action. If approved today, Quilience would be the only partial orexin 2 receptor agonist approved by the FDA, as well as the
only triple monoamine reuptake inhibitor approved by the FDA for the treatment of narcolepsy. Narcolepsy is caused by a profound loss
of orexin producing neurons and a partial orexin 2 receptor agonist may help to replace the missing endogenous orexin peptide, addressing
the underlying orexin deficiency and reduce disease specific symptoms. In addition, its unique dual mechanism of action as also a monoamine
triple reuptake inhibitor further acts to reduce disease specific symptoms, offering patients a treatment option that may address simultaneously
and in concert the two primary symptoms of narcolepsy. |
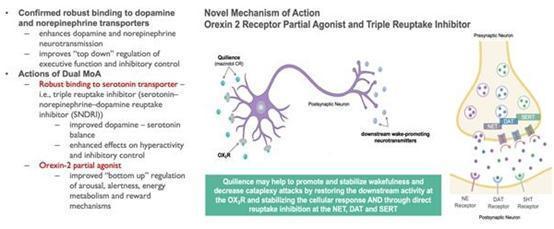
| ● | Low
potential for abuse and misuse and diversion. Mazindol is still listed as a Schedule IV controlled substance as classified by
the DEA. The DEA defines Schedule IV controlled substances as those “with a low potential for abuse and a low risk of dependence.”
Unlike sodium oxybate (a Schedule III controlled substance), the top-selling medication for narcolepsy in the United States with over
$ 2 billion in annual revenues, historically mazindol never required a REMS to manage known or potential serious risks associated with
its use. |
| ● | Quilience
is expected to be administered as a monotherapy. Narcolepsy is a complex spectrum disorder to manage and even with available
approved medications, the majority of narcolepsy patients often require multiple medications to treat their symptoms. According to the
current treatment guidelines (initially published in 2007) of the AASM, approved medications for narcolepsy, at best, provide only moderate
improvement in narcolepsy symptoms and their respective side effects may limit their use. The AASM specifically highlights that future
investigations should be directed toward development of more effective and better tolerated therapies and primary prevention. The Voice
of the Patient report from the FDA’s patient-focused drug development initiative, published in 2014, concluded that, based on the
overall benefit-risk assessment of currently approved medications, there is a continued need for additional effective and tolerable treatment
options for patients with narcolepsy. A retrospective analysis (Nittur et.al, Sleep Med. 2013 Jan;14(1):30-6) showed that mazindol has
a long-term, favorable benefit/risk ratio in 60% of drug-resistant hypersomniacs, including a clear benefit on the two primary symptoms
of narcolepsy, EDS and cataplexy. |
| |
● |
Quilience is expected to have minimal drug interactions. Based on the results of five in vitro metabolism studies, Mazindol ER has a very low potential for drug interactions In the lab, mazindol did not interact with any of the enzymes that metabolize drugs in humans. Its metabolism was also not influenced by any well-known enzyme inhibitors or stimulants. One of its metabolites (representing less than 12% of mazindol) showed some interactions with a minor metabolic enzyme named P-glycoprotein (Pgp) and this interaction will be further investigated in humans. Nevertheless, mazindol showed significantly less interactions with metabolic enzymes than any other narcolepsy treatment. |
| |
● |
Quilience is being developed as a once-daily oral tablet administered in the morning upon wakening. Patients have identified a need for treatment options that are easier to take, dosed less frequently, do not disrupt night-time sleeping and provide full day coverage of symptoms. We believe that once-daily dosing with Quilience may address this need and may help improve patient compliance and adherence with treatment. Quilience utilizes our proprietary extended-release formulation and is being designed to optimize its pharmacokinetic and pharmacodynamic properties with a rapid onset of action and prolonged controlled therapeutic effect, allowing for a daily oral dose that effectively provides consistent and long-acting symptom control to uniquely meet the needs of patients. |
Leveraging our scientific
insights and direct hands-on clinical experience, we are developing compounds that we believe have innovative mechanisms of action with
positive therapeutic profiles and represent a differentiated treatment option to overcome the limitations of the current therapies. For
example, Quilience and Nolazol are agents that differ significantly from available treatments in their apparent dual mechanism of action.
In addition to their action as a triple monoamine reuptake inhibitor that blocks serotonin, norepinephrine, and dopamine transporters,
they also target the orexin pathway as a selective (in relation to the orexin-1 receptor) partial agonist of the OX2R. This activity could
provide cortical regulation of the lower brain, and thereby lead to improved attention and inhibitory control, while also improving regulation
of executive function and inhibitory control which would provide both “top down” and “bottom up” regulation of
the brain. Orexin neurons (located in a part of the brain known as the hypothalamus) are multi-tasking neurons that serve as key modulators
to several vital functions, including sleep-wake states, attention, feeding behavior, energy homeostasis, reward systems, cognition and
mood throughout the cerebral cortex, and as such, the orexin system is a central promoter of wakefulness. A drug that targets orexin could
potentially increase arousal in individuals with ADHD, which could be useful in individuals who have a low level of arousal or motivation,
as is not infrequently the case in individuals with ADHD. To the best of our knowledge, no currently available medication for ADHD or
for the treatment of EDS and cataplexy, the two main symptoms associated with narcolepsy, has effects on the orexin system. In addition
to narcolepsy and ADHD, we believe that our product compound, Mazindol ER, may have therapeutic potential across a range of diverse CNS
conditions such as RLS, IH, OSA, Kleine-Levin-syndrome, daytime sleepiness due to myotonic dystrophy Type 1 (DM1) and Prader Willi Syndrome,
given the mechanism of action.
Relationship Between Narcolepsy and ADHD
Narcolepsy and psychiatric
disorders have a significant but unrecognized relationship in which the two can coexist. However, narcolepsy is frequently misdiagnosed
initially as a psychiatric condition, contributing to the protracted time for accurate diagnosis and treatment. Narcolepsy is a disabling
neurological condition that carries a high risk for development of social and occupational dysfunction. Deterioration in function associated
with narcolepsy may lead to the secondary development of psychiatric symptoms and inversely, the development of psychiatric symptoms can
lead to the deterioration in function and quality of life. The overlap in treatments may further enhance the difficulty to distinguish
between diagnoses.
ADHD is the most common
neurobehavioral disorder characterized by symptoms of inattention, impulsivity and hyperactivity with an estimated prevalence rate
of approximately 4-12% worldwide, as reported by the paper, “Understanding Attention Deficit/Hyperactivity Disorder from
Childhood to Adulthood,” by Drs. Timothy E. Wilens and Thomas J. Spencer. On the surface, ADHD may appear to be the opposite
of narcolepsy; however, there may actually be significant clinical similarity between the two. Cumulative data about sleep problems
in children and adolescents with ADHD has shown that children with ADHD have had a higher rate of restless sleep, impaired sleep,
and daytime sleepiness than children without ADHD. However, it is unclear whether EDS in ADHD is due to nocturnal sleep disturbances
or primary vigilance disorders because shorter sleep onset latency is assessed by the Multiple Sleep Latency Test, or MSLT (an
objective physiologic measure of sleepiness), in ADHD, rather than in the control group irrespective of the presence/absence of
sleep disturbances.
On the other hand, problems
with sleep may represent an intrinsic component of ADHD. The presence of ADHD symptoms in children and adolescents with narcolepsy has
been found to be about two-fold higher than in the general control population and adults with narcolepsy have been found to have a much
greater likelihood of having a diagnosis of ADHD in childhood compared to the general control population. Hyperactivity seen in ADHD may,
in fact, be a compensatory response for individuals who are under-aroused or sleepy and ADHD symptoms contribute to poor quality of life
and increased frequency of depressive symptoms, similar to narcolepsy. To the best of our knowledge, almost all of the treatments used
in ADHD have mechanistic overlap with treatments used in narcolepsy for EDS and researchers suggest that the symptoms of EDS, fatigue,
and sleep fragmentation may be the cause for ADHD symptoms, which is consistent with similar findings in other hypersomnia disorders.
Business Strategy
Our goal is to continue building
a differentiated, global biopharmaceutical company that is patient-focused on the development of transformative therapies that address
critical unmet needs in rare and complex CNS and neurodevelopmental disorders, such as Central Disorders of Hypersomnolence (which include
narcolepsy as well as IH) and ADHD. As we navigate the competitive landscape of our industry, while focusing on development of our product
candidates, we also intend to continually seek out-licensing and asset sale transactions that we believe will allow us to drive greater
value for our shareholders. The key elements of our business strategy are to:
| ● | globally
develop Quilience for narcolepsy in adults (lead project); |
| ● | pursue
new indications beyond narcolepsy (e.g., Quilience in IH) (follow-on projects); and |
| ● | explore
expansion of our growing product pipeline either through in-house innovation or in-licensing. |
Our Portfolio and Lead/Follow-On Product Candidates
Quilience for Treatment of EDS and Cataplexy
in Narcolepsy (Lead Product Candidate)
Narcolepsy is a rare chronic
primary sleep disorder characterized by two main symptoms:
| ● | EDS:
This symptom is the inability to stay awake or alert throughout the day and, in general, EDS interferes with normal activities on a daily
basis, whether or not a person with narcolepsy has sufficient sleep at night. People with EDS report mental cloudiness, lack of energy
and concentration, memory lapses, depressed mood, and/or extreme exhaustion. |
| ● | Cataplexy:
This symptom consists of a sudden loss of muscle tone that leads to muscle weakness and a loss of voluntary muscle control, while being
fully conscious. It can cause symptoms ranging from slurred speech to total body collapse, depending on the muscles involved, and is
often triggered by intense emotions such as surprise, laughter, or anger. |
Nearly all patients diagnosed
with narcolepsy require lifelong treatment to combat the daily and debilitating symptoms, yet, most continue to suffer because a large
portion remain undiagnosed and/or because of inadequate treatments. One FDA approved medication for both EDS and cataplexy associated
with narcolepsy, sodium oxybate franchise (Xyrem/Xywav), although marketed with significant limitations impeding its use, generated revenues
of over $2.0 billion in 2022 according to Jazz Pharmaceuticals’ 2022 full year financial results issued on March 1, 2023. On October
15, 2024, CoherentMI published a report titled “Narcolepsy Market Outlook 2024–2031,” which estimated that the global
narcolepsy market would be valued at approximately US$ 2.53 billion in 2024, and projected it would grow to US$ 4.68 billion by 2031,
reflecting a CAGR of 9.3% over the forecast period.
As partial agonist of the
OX2R and a triple monoamine reuptake inhibitor, we believe that Quilience reflects a unique mechanism of action to address the shortcomings
of the approved medications and the unmet needs of patients. Quilience is being designed as an oral, once-daily treatment, intended for
long-term use. Through the use of mazindol in Quilience and the expected benefits of our proprietary controlled-release formulation, we
hope that regulatory agencies will conclude that Quilience has a desirable balance of efficacy, safety and tolerability to support marketing
authorization.
We also anticipate developing
additional mazindol based ER product(s) for broader hypersomnia indications, such as IH, where there is a critical unmet need and no currently
approved treatments available, in addition to other disorders, such as neurocognitive disorders implicated by EDS.
Quilience has been granted
orphan drug designation by both the FDA and European Commission for the treatment of narcolepsy, and if approved for marketing in adults,
this designation is expected to provide 7 years and 10 years of market exclusivity in the United States and Europe, respectively, and
with the potential for additional market exclusivity, if and when further developed and approved in pediatrics (extended for an aggregate
of 7.5 years and 12 years in the United States and Europe, respectively). Additionally, we have been granted formulation patents in several
countries including the U.S., Europe, Canada and South Korea for our proprietary ER formulation, which provide patent protection through
2037.
After obtaining an IND approval
mid-2021, we initiated a Phase 2 clinical trial in the third quarter of 2021 to evaluate Quilience as a once-daily monotherapy for the
treatment of EDS and cataplexy, the primary symptoms of narcolepsy. This proof-of-concept, or PoC, trial was conducted in approximately
20-25 specialized centers across the U.S. and positive top-line results were announced on September 27, 2022. On January 30, 2023, we
announced the completion of an open label extension study with Quilience(R) (Mazindol ER) for the treatment of narcolepsy. On March 27,
2023, we announced open label extension study six-month data for Quilience(R) (Mazindol ER) in the treatment of narcolepsy Type 1 and
Type 2.
As a result of the subsequent
open label extension, or OLE, interim data results discussed more fully below, we intend to pursue an expedited development program with
the FDA under the Breakthrough Therapy and Fast Track designation programs, as well as potentially a similar program, PRIME, with the
EMA. Collectively, these programs are designed to expedite the development and review of drugs intended to treat serious conditions and
fill an unmet medical need. We believe Quilience may qualify for these programs based on (i) positive real-world evidence, namely, previous
off-label use of Quilience’s active molecule, mazindol, in treating narcolepsy, which was prescribed under France’s Authorization
Treatment Use program for 17 years in patients who failed to respond or could not tolerate the available approved treatments, (ii) its
dual mechanism of action as a serotonin-norepinephrine-dopamine reuptake inhibitors, or SNDRI, and partial OX2R agonist, which we believe
are crucial in addressing the underlying symptoms of both EDS and cataplexy, providing a pharmacological profile targeting multiple CNS
pathways, and (iii) the limited availability of treatments that successfully treat both cataplexy and EDS in narcolepsy.
Additionally, we are planning
to leverage the demonstrated biological activity of the active molecule in Quilience to further expedite the clinical development, as
well as drawing on the data from our ADHD program to support our clinical development efforts of Quilience and the FDA’s previous
approval of mazindol (in its immediate release form in the pharmaceutical product “Sanorex®,” as manufactured and distributed
by Novartis (through its Sandoz division) as safe in the management of exogenous obesity through the 505(b)(2) regulatory pathway. In
March 2021, we entered into a license agreement with Novartis Pharma AG, whereby we obtained, on an exclusive basis in the U.S., all of
the available data referred to and included in the original NDA for Sanorex® (mazindol) submitted to the FDA in February 1972. The
agreement encompasses all preclinical and clinical studies, data used for manufacturing including stability and other chemistry manufacturing
and controls data, formulation data and know-how for all products containing mazindol as an active substance, and all post-marketing clinical
studies and periodic safety reports from 1973 onwards. Under the agreement, we have obtained the same rights on a non-exclusive basis
in all territories outside of the U.S. except for Japan, with the right to cross-reference the Sanorex® NDA with non-U.S. regulatory
agencies in the licensed territories.
We believe that previous off-label
use of Quilience’s active molecule, mazindol, in treating narcolepsy, which was prescribed under a regulatory authorized CUP in
France for 17 years in patients who failed to respond or could not tolerate the available approved treatments, supports our development
of Quilience. We believe this positive real-world evidence in improving both EDS and reducing cataplexy demonstrates the potential for
Quilience to be a treatment for patients suffering from narcolepsy, and notably, also the potential to provide an alternative treatment
for patients who have not had a successful outcome with approved treatments, such as sodium oxybate, modafinil, methylphenidate-based,
and amphetamine-based products.
In most patients, narcolepsy
is caused by the permanent loss of orexin (also called hypocretin), which has been identified as the major regulator in the brain that
modulates the stability of the sleep-wake cycle. Individuals with narcolepsy often emphasize that their symptoms go beyond falling asleep
during the day and report feeling chronically tired, fatigued, or sleep deprived and also struggle with cognitive difficulties that interfere
with their ability to perform at work or school and devastating cataplexy attacks that can be debilitating and frightening. These impairments
are associated with an increased risk of accidents, trauma, and difficulties in maintaining personal relationships. The economic burden
of narcolepsy is also extensive and people with narcolepsy have above-average rates of health care visits, medication use, and unemployment,
and those employed have lower income levels. According to the Narcolepsy Network, narcolepsy affects an estimated 1 in every 2,000 people
in the United States, which equates to 150,000 to 200,000 patients in the United States and 3 million worldwide; however, it is estimated
that only 25% of those living with narcolepsy have been diagnosed and are receiving treatment. Most people with narcolepsy begin having
symptoms in their teenage years, but the diagnosis is usually not made until adulthood, suggesting that narcolepsy is both an underdiagnosed
and an undertreated disorder. According to the August 2018 National Know Narcolepsy Survey, most narcolepsy patients remain unsatisfied
on current treatments.
While symptomatic improvement
is possible, patients’ needs are usually not met and the therapeutic effects of the currently approved treatments remain inadequate
for most patients, including lack of symptom control, variable efficacy, rebound sleepiness and rebound cataplexy, troublesome side effects,
inconvenience, and high potential for abuse. Given the considerable burden of the condition, the adverse effects on the health of narcolepsy
patients, and the limitations of available medications, there is a critical unmet need for additional treatment options. Quilience is
a triple monoamine reuptake inhibitor, a SNDRI and partial agonist of the OX2R that may mimic the natural sleep-wake process by activating
and further enhancing the brain mechanisms that promote and regulate wakefulness. We believe Quilience may bridge this considerable treatment
gap and that the current narcolepsy landscape may provide an opportunity to establish ourselves as a leader in this space.
Real-World Evidence in Narcolepsy
The use of real-world evidence
may improve the quality and efficiency of clinical development and clinical trial design, with the potential to accelerate the development
of therapies that may provide meaningful benefits to patients. Real-world evidence is the analysis of real-world data, which can originate
from sources such as CUPs, and may provide a more complete picture of patient experience to inform patient-focused drug development, and
support the advancement of randomized, placebo-controlled clinical trials to support a marketing application.
While no longer commercially
available in the United States or Europe, the active molecule in Quilience, mazindol, was previously widely used off-label and for several
decades for the treatment of narcolepsy. Additionally, it was prescribed under a long-term CUP administered and regulated by ANSM (“L’agence
nationale de sécurité du medicament et des produits de santé”), the national agency in France responsible for
overseeing the safety of medicines and health products, helping to address the unmet needs of patients who did not initially respond,
later failed or were unable to tolerate the approved available treatments, such as modafinil, methylphenidate, sodium oxybate, and amphetamine-based
products. This CUP ran for 17 years, ended in 2016 due to unavailability of the product, and more than 200 patients with narcolepsy who
were refractory to available treatments were prescribed mazindol for the treatment of EDS and/or cataplexy.
A retrospective, multi-center,
observational study of this real-world data, financed by the French Health Ministry, and conducted in part by certain members of our team
and members of our Scientific Advisory Board (prior to their work with us), was performed to evaluate the effectiveness and safety of
mazindol in real-world, clinical practice. A total of 94 patients with narcolepsy, and suffering from cataplexy, including adults and
children, with a mean 30 months of treatment exposure were included in the analysis to evaluate the effectiveness of mazindol on improving
EDS. Mazindol noticeably improved EDS (p<0.0001), as measured by the ESS, a validated patient-reported measure of the patients’
recent likelihood of falling asleep in everyday activities and is the same primary outcome measure widely used in other narcolepsy-related
Phase 3 clinical trials. An analysis was also performed assessing the effectiveness of mazindol in controlling cataplexy in 62 patients
and demonstrated a statistically significant reduction in the frequency of cataplexy episodes (p<0.0001). The ESS score before mazindol
was 18.0 ± 3.1 and decreased to 13.6 ± 5 after mazindol, for a change of -4.2 (p<0.0001). In addition, the change in
weekly cataplexy rate before and after mazindol changed from 4.6 ± 3.1 (before mazindol) to 2.0 ± 2.8 after mazindol (for
a change of -2.7) (p<0.0001).
This retrospective study with
analysis of real-world data, provides positive real-world evidence that the treatment was well-tolerated and effective, in terms of improvement
in EDS and the reduction in cataplexy events, with a conclusion of an overall positive benefit-risk ratio in the majority of patients.
In their report concluding the results of the study (Nittur et.al, Sleep Med. 2013 Jan;14(1):30-6), the researchers found that Mazindol
has a long-term, favorable benefit/risk ratio in 60% of drug-resistant hypersomniacs, including a clear benefit on cataplexy and that
the data, taken altogether, suggest that mazindol has a major effect on sleepiness, possibly greater than that of modafinil (Provigil).
Narcolepsy Overview and Market Opportunity
Narcolepsy is the inability
to stay awake and arises from the dysregulation of the sleep-wake cycle, and in most individuals is thought to be caused by an immune
system mediated destruction of brain cells that contain orexin. Orexin is a neuropeptide and is critical to the maintenance of wakefulness,
continuity of sleep and coordination of the timing and features of REM sleep. Narcolepsy is often debilitating and incapacitating for
those affected and with no known cure, it usually requires life-long symptomatic treatment. Narcolepsy symptoms significantly interfere
with and cause a negative impact on cognitive, psychological and social functioning and can greatly affect daily activities.
The clinical hallmark of narcolepsy
is EDS, which is present in all narcolepsy patients and is generally the first symptom to occur. EDS manifests as sudden irresistible
bouts of sleep throughout the day, which can strike at any time, leading to inadvertently falling asleep during a variety of situations,
such as while at work or at school, when having a conversation, playing a game, eating a meal, or, most dangerously, when driving or operating
other types of machinery. A prominent and distinctive symptom of narcolepsy is cataplexy, which occurs in approximately 70% of those patients
with narcolepsy. Although the severity of cataplexy is variable, it can be very frightening and usually causes additional disability.
In addition to cataplexy, individuals with narcolepsy may also have symptoms indicative of altered REM sleep. REM sleep is normally characterized
by dreaming and muscle paralysis that prevents an individual from acting out their dreams. In narcolepsy however, REM sleep can occur
at any time of day and elements of REM sleep can mix into wake, manifesting as cataplexy, hallucinations that occur when going to sleep
or upon waking, and sleep paralysis, a transitional state between wakefulness and sleep in which one is aware but cannot move, speak,
or react.
Narcolepsy is classified into
two subtypes, narcolepsy type 1 and narcolepsy type 2. Type 1 is characterized by cataplexy or an undetectable or low level of orexin
in the cerebrospinal fluid, or CSF. In type 2, patients generally have less severe symptoms and do not experience cataplexy attacks. The
underlying cause of narcolepsy type 2 is unknown and many patients have normal CSF orexin levels; however, 25% of patients with narcolepsy
type 2 do have intermediate CSF orexin levels and these individuals are more likely to develop cataplexy and subsequently be diagnosed
with narcolepsy type 1.
Cataplexy is a symptom
that occurs almost exclusively in people with narcolepsy type 1, so its presence strongly suggests this diagnosis. However,
cataplexy can sometimes be subtle or mild, making additional testing sometimes necessary, with the diagnosis depending on ruling out
causes of secondary hypersomnia and performing an overnight sleep test with MSLT. On the MSLT, it takes less than eight minutes on
average for people with narcolepsy type 1 to fall asleep, and REM sleep is observed on at least two naps. People with narcolepsy
type 2 show the same results on MSLT as those with cataplexy.
In addition to EDS, cataplexy
and other symptoms of altered REM sleep, people with narcolepsy type 1 are prone to gain significant weight, which may be due to the loss
of orexin. Soon after narcolepsy onset, children can rapidly gain 5-15 kg (approximately 11 to 33 pounds) and adults with type 1 are often
overweight or obese, despite normal caloric intake and activity levels. Given that the active molecule in Quilience was previously approved
for the management of exogenous obesity and its multifunctional mechanism of action targets two pathways known to be implicated in obesity,
Quilience may also help to counter this negative effect of weight gain associated with narcolepsy.
Recently, we received a granted
patent (U.S. Patent No. 11207271), from the U.S. Patent and Trademark Office which covers oral formulations containing immediate-release
and sustained-release layers of mazindol and their use in the treatment of attention deficit disorders (ADD or ADHD), related deficit
of alertness or decline in vigilance, or EDS (e.g., narcolepsy, IH).
The patent has a term that
expires no earlier than April 2037. Based on its current clinical development plans to obtain regulatory approval, we intend to list the
patent in FDA Approved Drug Products with Therapeutic Equivalence Evaluations, or Orange Book, if it receives market approval.
In January 2023, we announced
the new in vitro study results demonstrating the agonist effect of Mazindol ER at the Orexin-2 Receptor (OX2R). Two identical studies
measuring differing concentrations of Mazindol ER confirmed significant OX2R partial agonist activity at 30μM or higher. Notably,
findings show that Mazindol ER showed strong OX2R partial agonist activity by cellular and nuclear receptor functional assays. Results
showed pEC50 (a logarithm measure of drug potency expressing a concentration that is effective in producing 50% of the maximal response)
values of 4.7 to 5 for mazindol on the OX2R, indicating a strong OX2R partial agonist. Additional pre-clinical in vivo studies are currently
underway to confirm OX2R activity. A pilot study in OXR2 KO mice animal model compared favorably with investigational drug, TAK-925, an
orexin 2 receptor agonist.
Prevalence
According to the European
Narcolepsy Network, narcolepsy affects only 20 to 40 out of every 100,000 people; however, it is estimated that only one out of four people
living with narcolepsy have been properly diagnosed. Although it usually has an early age of onset during the adolescent years, a diagnostic
delay that often exceeds 10 years from the time of symptom onset suggests that narcolepsy is both under diagnosed and under treated. This
delay may result from several factors, including lack of clinician and patient recognition of the signs and symptoms of narcolepsy and
leading to multiple physician visits before receiving a diagnosis, as well as misdiagnosis of narcolepsy as another condition, such as
epilepsy, depression, or ADHD, which further delays treatment.
Current Treatment Landscape and Treatment Limitations
Until a cure is available,
the current treatment of narcolepsy focuses on symptom control, with the goal of keeping the patient alert during the day, primarily by
improving EDS and minimizing the occurrence of cataplexy. More than 90% of individuals with narcolepsy require chronic use of medication
to manage symptoms, in order to allow for important everyday activities to be performed safely, such as attending school, going to work
and caring for a child.
The current available treatment
options require the careful balancing of the drug’s efficacy, convenience of administration, development of drug tolerance, adverse
effects, comorbidities, monitoring for evidence of drug abuse, and new life circumstances, such as school, pregnancy and parenthood. In
narcolepsy, several classes of drugs are used for the treatment of EDS, including a CNS depressive agent, wake-promoting agents, a histamine
3 receptor antagonist/inverse agonist, and CII controlled stimulants.
Sodium oxybate (commercial
brands: Xyrem (Jazz), Xywav (Jazz), Lumryz (Avadel)) is the legally manufactured form of gamma hydroxybutyrate, an illicit drug of abuse.
It was the first FDA approved treatment for cataplexy and is also approved for EDS, and as such, is often used as a first-line therapeutic.
While it has been reported to have a positive impact for patients, sodium oxybate also has many challenges with significant limitations
that can often impede its use. As a severe CNS depressant with a rapid onset of sedation with both hypnotic and amnestic effects, it is
abused to incapacitate victims for sexual assault. Sodium oxybate is a CIII controlled drug and with the potential for an adverse outcome,
it is further subject to higher CI controls for abuse and is only available through an FDA imposed restricted-access REMS program. It
carries a Black Box warning for respiratory depression and abuse, which can lead to seizures, decreased consciousness, coma and death,
and at doses lower than those used to treat narcolepsy. The occurrence of experiencing adverse effects is more common with sodium oxybate
compared to other medications used in narcolepsy and adverse effects, even at recommended doses, include nausea, confusion, CNS and respiratory
depression, neuropsychiatric depression and confusion, bed-wetting, sleepwalking, automatic behaviors, and involuntary movements. Sodium
oxybate has a short half-life and is administered in a split dose, once at bedtime and again two and a half to four hours later, which
can be difficult for patients to manage. In addition, generally, an extensive titration period is required, which can take upwards of
seven months to achieve a complete optimal response.
Many patients with narcolepsy
have cardiovascular risk concerns, including hypertension, and treatment with sodium oxybate contributes 1,100-1,640 milligrams, or mg,
to an individual’s daily sodium intake, in comparison to a total daily intake of 1,500 mg as recommended by the American Heart Association.
Additionally, life-style changes are also often needed when being treated with sodium oxybate, including the avoidance of alcohol and
other medications that may cause sedation and due to its profound sedation and hypotonic effects, a change in living arrangements may
be needed if living alone or the need to seek different and multiple treatment options when becoming a parent. Yet, despite these severe
limitations, sodium oxybate continues to be the market leader in the United States in terms of revenues.
Our Solution: Quilience for Narcolepsy -
A Well-Suited Approach for the Disease Pathology
Narcolepsy is a debilitating
neurological disorder and the currently available treatment options are not considered sufficiently effective for most patients. This
is highlighted by the results of the recent 2018 “Know Narcolepsy Survey,” conducted by Versta Research, that emphasizes
the continuing and substantial burden of narcolepsy with an astonishing 88% of patients indicating that their current treatments are not
effectively managing their symptoms, while 94% and 93% stated that new treatment options are needed and expressed frustration with current
treatment options, respectively.
Quilience has a mechanism
of action that is distinct from existing and emerging therapies and we believe that, if approved, Quilience may represent a substantial
improvement to existing treatments. Mazindol’s mechanism of action, which may restore orexin signaling in the brain and further
enhance monoamine availability in promoting wakefulness and reducing cataplexy has the potential to be a breakthrough treatment and thereby
offering a significant treatment advancement. Furthermore, in November 2019, the Swiss Narcolepsy Network endorsed Quilience as a potential
novel treatment of narcolepsy. The Swiss Narcolepsy Network stated that its decision is based on several decades of highly promising off-label
use and compassionate use of mazindol in patients with narcolepsy.
Quilience Label Expansion
Following our current
focus on the development of Quilience for narcolepsy in adults, and if approved for marketing, we intend to seek a label expansion
for the treatment of narcolepsy in pediatric patients, which may require additional nonclinical and clinical studies. We are also
aiming to develop Quilience for the treatment of IH, a rare and chronic hypersomnia disorder for which there is currently no
effective or approved treatments available. Its hallmark symptom is chronic EDS and a craving to sleep during the day, regardless of
how many hours slept at night, which results in such persons taking daytime naps that are usually long and not refreshing.
Individuals with IH struggle to wake, despite setting multiple alarms and may have difficulty rising from bed, called sleep inertia.
Sleep inertia also includes feelings of grogginess upon waking and can result in impaired alertness and interfere with the ability
to perform mental or physical tasks. Similar to narcolepsy, people with IH may also suffer from hallucinations and sleep paralysis
when going to bed or upon waking.
The active molecule in Quilience
was also prescribed under compassionate use for the treatment of IH, providing positive real-world evidence of its benefit in improving
EDS specifically in patients with IH. We have received orphan drug designation from both the FDA and the European Commission for the treatment
of IH and this designation is expected to provide us initially with 7 years and 10 years of market exclusivity in the United States and
Europe, respectively. Additionally, the recently granted patent for a proprietary modified-release formulation, provides patent protection
through 2037 in the United States.
Quilience Development Program
We conducted a Phase 2 randomized,
double-blind, placebo controlled clinical trial in adult patients with narcolepsy in the third quarter of 2021 and concluded the trial
in the third quarter of 2022. The primary endpoint of the study was the change from baseline in EDS, as measured by the Epworth Sleepiness
Scale (ESS). The key secondary endpoint was the change from baseline in the weekly number of cataplexy attacks in the subset of patients
with cataplexy. The trial design is shown below:


The trial was conducted in
21 clinics in the U.S. and enrolled 67 patients with both narcolepsy Type 1 and Type 2, who received treatment with either 3mg of Quilience
(Mazindol ER) once daily or placebo. Patients in the trial were randomized 1:1 into each treatment arm. All patients who completed the
Phase 2 POLARIS study were eligible to participate in an open label extension (OLE) study and continued once-a-day treatment with Mazindol
ER 3 mg for up to six months as monotherapy (no concomitant wake-promoting or anti-cataplexy treatments were allowed). On January 30,
2023, we announced that 87% of patients who completed the POLARIS Phase 2 study requested to continue monotherapy treatment with Mazindol
ER in the six-month OLE study rather than transition to other therapies.
In September 2022, we announced
top-line data relating to our Phase 2 POLARIS study for the use of Mazindol ER in patients with narcolepsy. Of the 60 patients targeted
for enrollment in the U.S. Phase 2 trial (Study NLS-1021), 67 were randomized and included in the final analysis. The final database included
33 patients on treatment and 34 patients on placebo, with balanced mean baseline ESS scores (17.9 for treatment and 18.0 for placebo).
Approximately one-third of patients enrolled in the study were diagnosed with narcolepsy type 1 (NT1), and therefore presented with both
EDS and cataplexy symptoms. Eligible NT1 patients must have had moderate to severe disease according to the study protocol - defined as
having more than 3-4 cataplexy attacks per week. Study participants were required to undergo a 1-2-week wash out period (depending on
prior therapy). After the wash out period, participants were randomized to receive either once-daily treatment with Mazindol ER 2mg for
week 1 and 3mg for weeks 2-4, or matching placebo for 4 weeks.
For EDS, the ESS mean change
from baseline to each visit and the standard error (SE) of Quilience® (NLS-2) versus placebo were all statistically significant at
week 1 -4.3 (1.13) versus -1.1 (1.06) (p=0.0055), at week 2 -4.7 (1.14) versus -1.3 (1.06) (p=0.0035), at week 3 -5.0 (1.18) versus -1.6
(1.09) (p=0.0045), and at week 4 -5.8 (1.23) versus -2.1 (1.14) (-6.26, -1.15; p=0.0045).
Observed Mean Change from
Baseline in ESS:
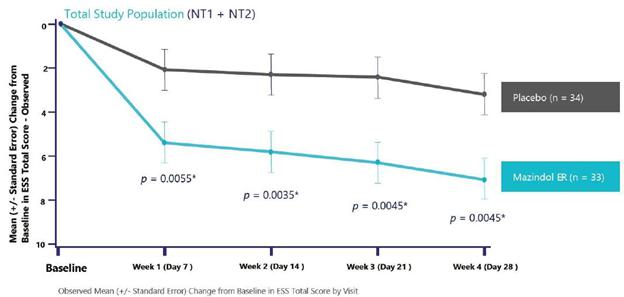
The change from baseline to
Week 4 in the weekly average number of cataplexy episodes decreased over time in both treatment groups. The change from Baseline to Week
4 in Average Weekly Cataplexy Episodes was statistically significantly greater in the Mazindol ER group (mean [SD] –14.3 [9.50])
than in the placebo group (–6.1 [6.30]) (p = 0.019) despite the smaller sample size (only 1/3 of the study population were NT1 patients).
Observed Mean Change from
Baseline in Weekly Cataplexy Episodes:
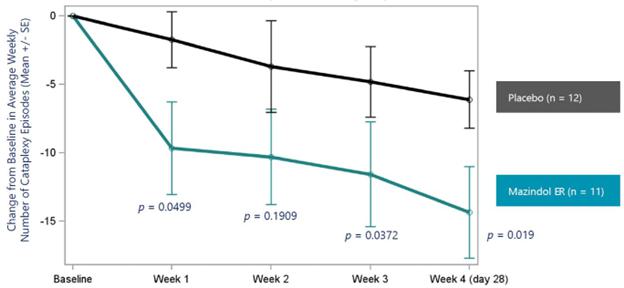
The treatment arms were balanced
in terms of patient demographics, baseline levels, and disease characteristics. Mazindol ER was well-tolerated and no safety concerns
were identified. No serious adverse events were reported.
Mazindol ER was generally
safe and well-tolerated in the completed Phase 2 trial. There were 66 patients in the Safety Population (all patients who received at
least one dose of Mazindol ER). Treatment emergent adverse events (TEAEs), were reported by 48.5% (16/34) of patients receiving Mazindol
ER and 23.5% (8/33) of patients receiving placebo. There were no unexpected adverse events. There were no severe and no serious TEAEs
reported for either the placebo or the Mazindol ER groups. In addition, no patients discontinued Mazindol ER due to TEAEs. The most common
TEAEs for the Mazindol ER group was dry mouth (21.2% [7/34]) followed by nausea (9.1% [3/34]), and decreased appetite (9.1% [3/34]). The
most common TEAEs for the placebo group were dry mouth (2.9% [1/34]), urinary tract infection, (2.9% [1/34]), and headache (2.9% [1/34]).
Other than an expected reduction in body weight (~1.3 kg) and a moderate increase in heart rate (~11.3 bpm), there were no other significant
ECG, labs, or vital signs changes on mazindol versus placebo.
Patients who completed
the POLARIS Phase 2 trial were able to participate in the OLE study. The OLE study enabled patients completing the randomized controlled
trial to access treatment with Mazindol ER without any background stimulant and/or anti-cataplexy treatment for up to 6 months. Of the
67 patients who completed the randomized controlled Phase 2 trial, 52 patients (or 87% of completers) elected to roll over into the OLE
study. Patients treated with Mazindol ER in the randomized Phase 2 trial continued to improve after rolling over into the OLE study. Placebo
patients in the randomized Phase 2 trial achieved comparable results to treated patients in the Phase 2 trial after switching to Mazindol
ER in the OLE study. Safety and tolerability of Mazindol ER were similar between the randomized trial and OLE study.
For patients treated with
Mazindol ER in the randomized Phase 2 trial, ESS improved by an additional 1.8 points in the OLE study by the third month of treatment
(double-blind and OLE treatment combined). At that timepoint, the mean ESS score for these patients reached 8.2, with lower scores denoting
an improvement in the condition (improved wakefulness). Importantly, ESS scores of 10 or below are considered to be typical scores for
people without narcolepsy. As an extension of the 4-week randomized treatment period in the Phase 2 trial, these data indicate that maximum
efficacy for EDS with Mazindol ER is reached at approximately 3 months of treatment. Overall, the mean score for these patients declined
by approximately 10 points from their baseline levels at the start of the randomized Phase 2 trial to month 3 in the OLE study.
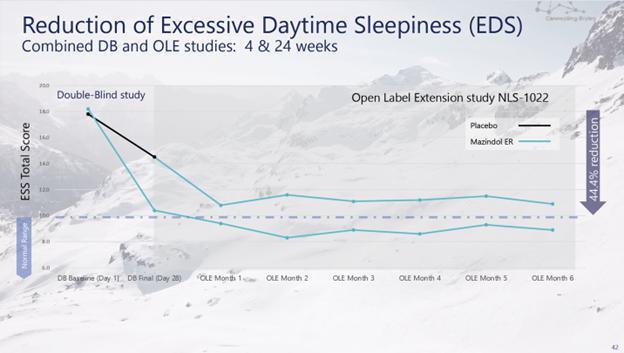
For patients who received
placebo in the randomized Phase 2 trial and rolled over to receive Mazindol ER in the OLE study, EDS scores declined to levels comparable
to those treated with Mazindol ER in the randomized trial at Week 4. This effect was maintained through month 3 in the OLE study, with
EDS scores just above the “normal” range.
For patients diagnosed with
cataplexy and treated with Mazindol ER in the randomized Phase 2 trial, the mean number of weekly cataplexy episodes was approximately
10 at the end of the 4-week DB period, down from a baseline level of approximately 17.5 at the beginning of the trial. During the OLE
study, mean weekly cataplexy episodes for these patients declined to 2.1, and remained relatively stable in the 2 to 4 range through week
24 of the OLE study.
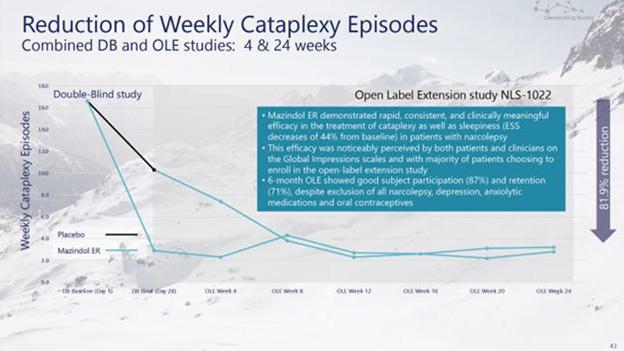
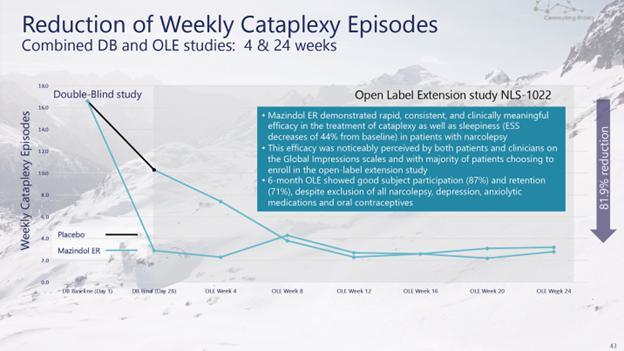
Notably, there were patients
diagnosed with Narcolepsy Type I who achieved zero weekly cataplexy episodes in the OLE study, with some of those maintaining this effect
through week 12 and beyond. Below is an example of one of those super responders:
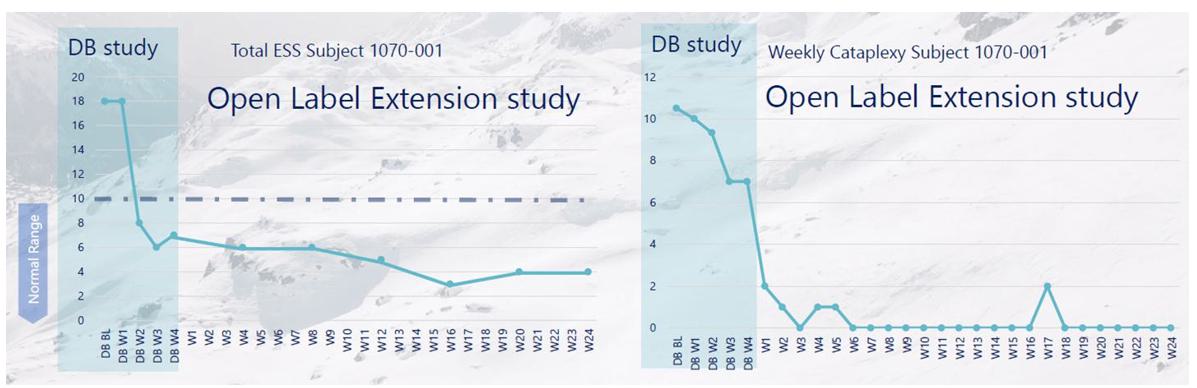
Given the success of our Phase
2 clinical trial, we intend to commence two identical, placebo-controlled Phase 3 trials in adult patients with
narcolepsy type 1. On March 29, 2023, we met with the FDA for an end of Phase 2 meeting. On May 2, 2023, we announced that the FDA provided
authorization to proceed with the Phase 3 clinical program (AMAZE) for Mazindol ER. In July 2023, we announced that the first Phase 3
clinical trial protocol received approval from the independent IRB. The AMAZE Program encompasses two almost-identical double-blind Phase
3 studies (N=50 each) investigating Mazindol ER versus placebo in adult patients with narcolepsy. Along with IRB approval and the green
light from the FDA, NLS has retained a CRO and has enrolled a number of sites for phase 3 studies. Once suitable capital has been
secured, we expect the phase 3 program to commence as the sites are ready to begin enrolling patients.
Additional clinical studies
may be required for regulatory approval necessary to commercialize Mazindol ER, such as clinical pharmacology studies, and, if needed,
we intend to conduct these studies in parallel with our Phase 3 program, subject to agreement with the FDA and other applicable regulatory
authorities.
Both phase 3 trials, NLS-1031
and NLS-1032, will measure the weekly cataplexy episodes as the primary endpoint over eight weeks of treatment. Patients will then continue
into a 12-month OLE phase of each study. To be eligible for enrollment into the program, patients must be at least 18 years of age and
have been diagnosed with narcolepsy with cataplexy.
As agreed with the FDA, we
intend to submit the NDA for Quilience as a new NDA. Nevertheless, a large amount of the original NDA data will be used for this submission.
To license the original mazindol data, we entered into an agreement with Novartis in March 2021 to obtain all preclinical and clinical
data for studies previously conducted on mazindol by Novartis. This data may potentially provide us another avenue to streamline and/or
reduce the costs of our preclinical and clinical programs. Our ability to rely on the FDA’s previous findings of safety studies
published in the scientific literature, and the extent to which licensed innovator data may be utilized will depend on our ability to
demonstrate a scientific bridge to Mazindol ER from the previous formulation.
In November 2022, we launched
an individual Paid-for Named Patient Program, or NPP, to provide access to Mazindol ER for the treatment of IH in Europe where this medication
would not otherwise be available for this indication in certain countries. The NPP for IH was launched in the United Kingdom and we were
expected to expand to other countries including France, Italy and Switzerland. This NPP was terminated in May 2023 for lack of performance
of Calcog (Caligor Coghlan Ltd.), the service provider in charge of the development and execution of this program in Europe. We partnered
with a third-party pharmaceutical company to expand access, early access and compassionate use programs to provide treatment of IH with
Mazindol ER where it would otherwise not be available.
In January 2023, we announced
a technology patent grant covering Mazindol ER for treatment of ADHD and IH in Hong Kong. In October 2024, we announced our patent grant
covering Mazindol ER for the treatment of heroin dependence in Japan. This further supports our global strategy with key patents granted
the following major markets: Hong Kong, Japan, South Korea, the U.S., Europe & Canada.
Compassionate Use Objectives in CDH1
in Europe
While “compassionate
use” originally was understood to imply that the drug product is supplied for free, there is a growing understanding with policymakers,
payers and patient organizations, that paid-for CUPs are justifiable. In Europe and other regions of the world (excluding the United States),
it is recognized that CUP funding can provide an early source of biopharma revenues and incentivize the increased availability of potentially
transformative treatments, especially for rare disorders and orphan diseases. Given the extensive therapeutic experience with mazindol,
including off-label in narcolepsy patients, we believe that there is ample precedence to justify a funded CUP in selected European countries
for a defined group of patients.
We intend to conduct either
or both a cohort program and a named patient program, or NPP, each of which is highlighted below in a variety of European countries. Although
we have yet to determine which countries, we will initiate CUPs in first, our current plan is to target the following countries in the
following priorities (priority in each wave has yet to be determined):
| |
● |
First Wave: United Kingdom, Netherlands, Belgium, France, Italy and Switzerland; |
| |
● |
Second Wave: Latin America, the Czech Republic, Denmark and Spain; and |
| |
● |
Third Wave: China, Germany, Austria, Japan, Sweden and Taiwan. |
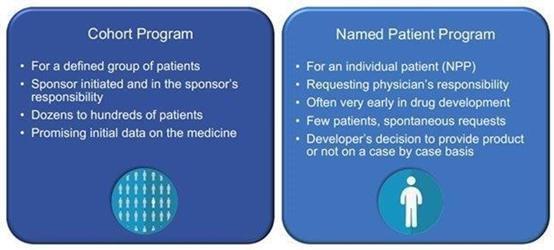
In addition to the potential
benefit of generating revenues before Quilience market authorization from the regulatory agencies, the collection of data within the CUP
is a key potential benefit for the overall Quilience evidence generation strategy, as we believe that a CUP program for Quilience would:
| ● | complement
the clinical data package for regulatory submissions; |
| ● | support
primary regulatory approvals with longer-term follow up effectiveness and safety data and patient lived experience evidence; |
| ● | support
label expansions in broader populations than included in the Quilience late state trials (e.g., in juvenile narcolepsy); |
| ● | potentially
accelerate regulatory approval in China if we were to seek regulatory approval for Quilience; |
| ● | enhance
the evidence package for market access and pricing and reimbursement; |
| ● | provide
qualitative live-experience data and information on the unmet needs from the patient (and caregiver) perspective that may contextualize
the net therapeutic benefits offered by Quilience within the narcolepsy management paradigm; |
| ● | bridge
patients from the end of Phase 2 and 3 clinical trials to commercialization (assure continued patient access); |
| ● | allow
us to generate hypotheses for label expansions in broader populations (as compared to clinical trials); |
| ● | allow
us to leverage real-world and patient-centric data collected (board populations, quality of life, satisfaction); and |
| ● | allow
us to professionally address patient and physician requests (formal published policy and process). |
| [1] |
Central Disorders of Hypersomnolence (including but not limited to narcolepsy type 1 / type 2, IH, etc.) |
In addition to these potential
benefits of a CUP program for Quilience, commercial objectives of such a program would include the ability to (i) generate pre-licensing
revenues in selected countries, (ii) receive post-authorization revenues in non-priority countries for commercial use, (iii) potentially
establish an early market presence with a key opinion lead, and center of excellence strategy and (iv) more rapid revenue uptake (than
a non-CUP program) upon commercial availability (patients already on product and treated).
Nolazol for the Treatment of ADHD (Back-up
Program Product Candidate)
Nolazol is a triple monoamine
reuptake inhibitor and orexin receptor-2 partial agonist and its unique pharmacological profile is expected to yield important benefits
compared to existing treatments of ADHD. Enhancing the function of the three neurotransmitters well-known to be implicated in ADHD, norepinephrine,
dopamine and serotonin, along with its activity on the orexin-2 receptor, Nolazol may produce an optimal reduction in ADHD symptoms over
available treatments.
Nolazol is supported by a
positive pilot clinical trial with mazindol in 24 children with ADHD and a positive Phase 2 clinical trial in 85 adults with ADHD. The
Phase 2 clinical trial in adults met all primary and secondary study endpoints and was well-tolerated. A robust effect on ADHD symptoms
was demonstrated with a large placebo-adjusted effect size of 1.09 in the investigator-rated ADHD symptom scores.
Although more than 25 different
products have been approved by the FDA since 1937 for the treatment of ADHD, many of which are no longer available, there still continues
to be a large treatment gap with no optimal treatment currently available. Physicians, patients and their caregivers press for improved
treatment options to address key shortcomings with currently available treatments, including the need for a more tolerable safety profile,
more consistent efficacy with no rebound effect, and the need for lower risk of abuse, dependence, and misuse. Currently, doctors, patients,
and caregivers must choose between treatments that may be effective, but come with significant safety liabilities, such as high potential
for abuse and risk of diversion coupled with tight restrictions on writing and filling prescriptions; or treatments that are unscheduled,
but are also typically less effective. We are seeking to develop Nolazol such that, if approved for marketing, it could be the drug product
to close this treatment gap and address this unmet medical need.
Given the positive outcome
of our Phase 2 trial in ADHD, we may initiate Phase 3 clinical trials if we receive FDA’s green light to proceed at a later stage.
ADHD Overview and Market Opportunity
ADHD is a chronic neurodevelopmental
disorder affecting children, adolescents and adults and is characterized by an ongoing pattern of inattention and/or hyperactivity-impulsivity
and is associated with clinically significant impairments in executive functioning. In addition, ADHD is one of the most commonly diagnosed
neurodevelopmental disorders in school-age children and it often persists into adulthood. It is characterized by symptoms of inattention
and/or hyperactivity-impulsivity, and affects cognitive, academic, behavioral, emotional, and social functioning. An estimated 8.4% of
children and 5% of adults in the United States have a current diagnosis of ADHD, and if left untreated can lead to poor occupational and
psychosocial outcomes. According to a report published by Global Market Insights on December 17, 2024, the global ADHD market was valued
at $15.8 billion in 2023 and is projected to grow at a CAGR of over 5.1% from 2024 to 2032. Furthermore, based on a report by Grand
View Research, Inc., ADHD is the 12th most prevalent therapeutic indication for dispensed prescriptions and is predicted to
grow at a 6.4% compounded annual growth rate until 2030. Despite CII classification and black box warnings and safety liabilities, Vyvanse,
Takeda’s branded ADHD medication, generated approximately $3.01 billion in global sales during fiscal year 2023, which ended on
March 31, 2024. In August 2023, Vyvanse lost market exclusivity in the United States, resulting in the introduction of multiple generic
alternatives. This loss of exclusivity contributed to a decline in sales, and Takeda has projected a further decrease of approximately
47% in Vyvanse revenue for fiscal year 2024 due to increased generic competition.. Furthermore, based on a report from LifeSci Capital,
it is estimated that CNS stimulants - amphetamine and methylphenidate containing products, represent approximately 90% of all ADHD drug
sales.
Across the lifespan, ADHD
may impede cognitive functioning as well as mental health and development and can have a significant social impact on patient’s
lives, causing disruption at school, work, and in relationships and can also be associated with risk-taking or criminal behavior and other
far-reaching effects on wider society. Over the past eight years, ADHD diagnoses have increased 30% in the United States, a trend indicating
the need to focus on the diagnosis and treatment for a growing number of patients.
The Centers for Disease Control
and Prevention, or the CDC, reported that 6.1 million, or 9.4%, of children and adolescents in the United States have ever been diagnosed
with ADHD and 5.4 million, or 8.4%, have a current diagnosis, and 62% take medication, while 47% receive behavioral therapy and 23% receive
no treatment at all. According to the CDC’s 2022 National Survey of Children’s Health, approximately 1 in 9 U.S. children
(11.4%, or 7.1 million) have been diagnosed with ADHD at some point, and 10.5% (6.5 million) currently have the condition. Among those
with current ADHD, 58.1% have moderate or severe symptoms, and 77.9% have at least one other co-occurring disorder. Roughly half (53.6%)
are being treated with medication, while nearly one-third are not receiving any ADHD-specific treatment at all. In terms of treatment,
53.6% were receiving medication, 44.4% had received behavioral therapy in the past year, and nearly one-third (30.1%) were not receiving
any ADHD-specific treatment. Between 2016 and 2022, the number of U.S. children diagnosed with ADHD increased by more than one million,
reflecting a significant rise in prevalence.. Additionally, ADHD is the second most impactful condition affecting children and adolescent
health in the United States, as measured by the Blue Cross Blue Shield Health Index, and children diagnosed with ADHD struggle with paying
attention, controlling impulses and being overly active. Social skills in children with ADHD often are significantly impaired. Problems
with inattention may limit opportunities to acquire social skills or to attend to social cues necessary for effective social interaction,
making it difficult to form friendships. Hyperactive and impulsive behaviors may result in peer rejection. The negative consequences of
impaired social function, such as poor self-esteem, increased risk for depression and anxiety, may be long standing.
Once believed to only affect
children and adolescents, ADHD is now well understood to be a lifespan disorder that persists into adulthood in up to 65% of patients
affecting 1 out of 30 adults worldwide, as disclosed by the ADHD Institute, an educational platform developed and funded by Takeda, and,
based on our own assessments of data from the U.S. census Bureau, there are approximately 11 million adults in the United States with
ADHD. Research firm GlobalData reported that since 2015, the adult ADHD market has become larger and begun growing at a faster rate than
the pediatric ADHD market. Adult ADHD is often characterized by recurrent problems with restlessness, impulsivity, problems with time
management and finances, as well as problems regulating emotions. Rather than being hyperactive like children, adults with ADHD report
experiencing an internal sense of fidgetiness and restlessness and with signs of inattention more apparent through problems communicating
with others. Upon entering the job market, many adults also experience obstacles in employment, and are at increased risk to be terminated
due to repeated tardiness or absenteeism. Such difficulties contribute to poorer employment outcomes and a lower likelihood of being employed
in professional environments. Adults with ADHD often find it difficult to generate effective solutions to social problems and these deficits
in social cognition can increase the likelihood of peer rejection, and social isolation, adding to struggles with depression and social
anxiety.
Throughout an individual’s
lifetime, untreated ADHD can increase the risk of psychiatric disorders, educational and occupational failure, accidents, criminality,
social disability and addictions and ADHD treatment is usually initiated with stimulants, such as amphetamine and methylphenidate-based
products. While these drugs may provide a generally effective treatment option for patients, they also have serious safety concerns and
are misused recreationally with both diversion and abuse being a common and significant risk. As a result, they are controlled substances
and classified under the CSA as CII stimulants. Consequently, they all carry an FDA imposed Black Box warning on the drug label to call
attention to these serious or life-threating risks. Further, not all individuals respond optimally to or can tolerate CII stimulants and
their use is contraindicated in numerous patients, including those with tics, anxiety and other certain psychiatric disorders, cardiovascular
concerns, substance use disorder, or stimulant refusal. A few non-stimulant treatment options are available; however, their efficacy is
not as robust, and their tolerability profile is not necessarily improved when compared to CII stimulants. In addition, the few non-stimulant
treatments available are generally considered as second-line treatments and are often used in conjunction with schedule II (CII) simulants
rather than as a therapy that uses one type of treatment, commonly referred to as a monotherapy.
The availability of a treatment
that has robust efficacy on par with CII stimulants and that is well tolerated with lower potential for abuse represents what we believe
is an important unmet need to persons with ADHD. The magnitude of the treatment effect demonstrated in our Phase 2 study is comparable
to what is typically seen with the leading CII stimulants, and, mazindol, the active molecule in Nolazol, is currently a CIV controlled
substance under the CSA, meaning it has an already established low risk of abuse. We believe Nolazol, whose active ingredient is mazindol,
a CIV stimulant, may be the transformative treatment for ADHD that physicians and patients have been waiting for, by providing a treatment
with what we believe will be similar efficacy as CII stimulants, but with a low potential for abuse.
We have a robust method of
use patent for Nolazol for the treatment of ADHD expiring in August 2028 in the United States and in December 2027 in Europe. Additionally,
we have filed an international patent application under the PCT, for a proprietary controlled release formulation, and, if granted, may
provide patent protection through 2037 in the United States.
Diagnostics and Patient Sub-Types
Healthcare providers use the
guidelines in the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, or DSM-5,
to help diagnose ADHD and this diagnostic standard helps ensure that people are appropriately diagnosed and treated for ADHD. The DSM-5
identifies three sub-types and each presentation is distinguished by a distinct set of symptoms that physicians use to diagnose the condition.
The three presentations are: (1) Predominantly Inattentive; (2) Predominantly Hyperactive-Impulsive; and (3) Combined Presentation.
The diagnostic evaluation
for ADHD includes a comprehensive medical, developmental, educational, and psychosocial evaluation. This comprehensive evaluation is necessary
to confirm the presence, persistence, pervasiveness, and functional complications of core symptoms, exclude other explanations for core
symptoms and identify coexisting emotional, behavioral, and medical disorders. In order to meet criteria for ADHD, core symptoms must
also impair function in academic, social, or occupational activities.
Two-thirds of individuals
with ADHD have at least one other neurodevelopmental, psychiatric, or other CNS disorder, including anxiety, depression, autism, and sleep
disorders. Substance use disorder is a common comorbidity associated with ADHD and may have a direct underpinning in the pathophysiology
of the disease, amplifying the need for treatment options with a lower risk of abuse.
Adults with ADHD, with a delayed
diagnosis in adulthood, are often diagnosed after several attempts to find treatment for comorbid disorders, such as depression, substance
abuse, sleep disturbances, or anxiety. The presentation in adults is typically related to problems with work, disorganization, and tendency
to procrastinate, as well as anxiety, sleep disorders, and impulsivity.
We believe Nolazol could provide
a transformative treatment option for all individuals with ADHD and further could draw substantial market share from patients who have
failed or couldn’t tolerate stimulants; current patients, or their parents, with concerns with stimulant use; current patients seeking
the convenience of a CIV product compared to a CII product, allowing for generally no limitations on quantity, the ability to refill,
phone-in prescriptions, and less frequent office visits; individuals where abuse and/or diversion is a prominent concern; and individuals
or parents of individuals diagnosed with ADHD who have avoided treatment due to stimulant concerns and the associated stigma.
Current Treatment Landscape and Treatment Limitations
Although there is no cure
for ADHD, medications may help to reduce symptoms and improve functioning. The current treatment options for ADHD can be broadly classified
as either amphetamine or methylphenidate-based stimulants or as non-stimulants. Based on data
that we have collected, we believe that stimulants represent a majority of the ADHD drug market in the United States, with a market share
of approximately 90%.
Amphetamine and methylphenidate-based
products are all classified under the CSA as CII stimulants, due to their high potential for abuse and the risk of severe psychological
or physical dependence. These drugs are heavily controlled under U.S. federal and state laws, and are subject to criminal sanctions for
abuse, diversion and misuse and require a CII level prescription, and despite being a chronic disorder and need for daily medication,
this limits the quantity to a 30-day supply and is also not refillable. Consequently, all CII stimulants contain Black Box warnings, similar
to narcotics such as fentanyl and oxycodone, which are also CII substances. In addition, CII stimulants have the potential for numerous
adverse effects, as indicated by their warnings of serious cardiovascular reactions such as sudden death, stroke, and heart attack, psychotic
or manic symptoms in patients with no prior history, and are associated with peripheral vasculopathy, including Raynaud’s phenomenon.
CII stimulants are also sleep-affecting drugs and shorten total sleep time, increase the time it takes to fall asleep, adversely impact
the ability to stay asleep, and increase daytime sleepiness.
The long-term use of prescription
stimulants has been widely reported to cause drug tolerance, which is the loss of efficacy over time and requires a complete change in
treatment, or an increased dose of the existing treatment, or the add-on of another medication to the existing treatment in order to achieve
therapeutic effectiveness. This leads to an increased risk of developing serious adverse effects that are unrelated to ADHD. Another concern
in treating patients with CII stimulants is the potential “rebound effect” that occurs when the medication wears off, resulting
in the return of ADHD symptoms and which may also occur in an amplified form. In children especially, this often triggers increased irritability
and/or aggressive behavior and the rebound in children and adults may be exacerbated by multiple drug administrations, often used to obtain
the desired duration of effect or to address drug tolerance. Additionally, studies have highlighted that primary limitations of CII stimulants
are intolerable adverse effects that interfere with patient adherence rates and sub-optimal efficacy with the onset of drug tolerance.
According to the 2002 practice
parameter for the use of stimulant medications from the American Academy of Child & Adolescent Psychiatry, approximately 30% of patients
do not respond adequately to or have dose-limiting adverse effects with CII stimulants. Additionally, certain patients, or parents of
patients, prefer not to use CII stimulants due to their stigma and known abuse potential. There are a few non-stimulant treatments available,
such as atomoxetine (Strattera®), clonidine (Kapvay®), and guanfacine (Intuniv®), that were
developed to address this need; however, their efficacy is sub-optimal to stimulants and while unscheduled, their overall safety profile
does not necessarily provide an improvement to CII stimulants.
Strattera, a norepinephrine
reuptake inhibitor, was the first non-stimulant treatment option for ADHD and while its initial launch started strong, underscoring the
demand for an alternative to CII stimulants, sales steadily declined as patients and physicians found it to not be nearly as effective
as CII stimulants. It is now considered a second-line treatment and is typically used as an alternative to CII stimulants for patients
who have a substance abuse problem, a family member(s) with a substance abuse problem, tics, or intolerable side effects with CII stimulants.
Strattera carries a Black Box warning for increased risk of suicidal thoughts in children and adolescents and additional warning statements
for liver damage. Moreover, Strattera takes four weeks to reach initial onset of action and six to ten weeks to achieve full clinical
effectiveness, related both to the prolonged titration needed and the delay in the onset of action of the compound. Today Strattera (branded
and generic version) has about 3.6% market share, despite initially climbing to nearly 20% following launch on the basis of being an alternative
to CII stimulants.
Clonidine and Guanfacine are
also non-stimulants that are alpha-2 adrenergic receptor agonists and were both initially approved for managing blood pressure. They have
been approved for use in children and adolescents, generally in conjunction with CII stimulants as an add-on therapy. While they are used
in children and adolescents, there is little study of their efficacy and safety in adults. As a monotherapy, they are usually reserved
for children and adolescents who respond poorly after several trials of stimulants and Strattera, have unacceptable side effects with
stimulants or Strattera, or have significant comorbid conditions limiting the treatment options to these products. These two drugs have
not had significant commercial success, with a combined peak market share of about 5%.
Our Solution: Nolazol - Next-Generation ADHD Therapeutic
We believe a large market
opportunity exists for a non-CII stimulant, such as Nolazol, that uses mazindol controlled release as its active ingredient; mazindol
has been classified as a CIV stimulant, due to its low risk of abuse and tolerance.
Given its unique binding profile
(specifically, as a partial OX2R agonist), in addition to its classification as a CIV stimulant, we believe Nolazol could be transformative
for the ADHD treatment landscape. Nolazol is a triple monoamine reuptake inhibitor and also a partial agonist of the OX2R, which we believe
is an important, unique and differentiating factor relative to other ADHD treatments. In our clinical studies conducted to date, Nolazol
has been well-tolerated and there were no treatment-related serious adverse effects or discontinuations. In terms of abuse potential,
Nolazol has an already established low risk of abuse, as previously determined by the DEA when mazindol, its active ingredient, was scheduled
as a CIV substance, underscoring the awareness and agreement that Nolazol has a lower risk of abuse than CII stimulants. In addition,
based on the current DEA classification of mazindol as a CIV stimulant, we expect that Nolazol will continue to be without a Black Box
warning in the U.S., which is another important differentiator relative to both CII stimulants and the current non-stimulants in use today
to treat ADHD.
Our Phase 2 trial showed significant
improvement in ADHD symptoms, met all primary and secondary study endpoints and was well-tolerated and with no clinically significant
adverse effects over placebo. In light of its innovative mechanism of action and low potential for abuse, we believe Nolazol, if approved
for marketing, could represent a highly differentiated alternative to the CII treatments in use today to treat ADHD.
Nolazol Clinical Trial Results
Phase 2 Clinical Trial
We completed a Phase 2 clinical
trial in 2017 in the United States, in which Nolazol was well-tolerated and demonstrated statistically significant improvement over placebo.
The clinical trial met the primary and all secondary endpoints and had a robust effect on ADHD symptoms with a large placebo-adjusted
effect size of 1.09 in the investigator-rated ADHD symptom scores. We believe that the magnitude of this effect is comparable to currently
leading CII stimulants and considerably larger than the available non-stimulant treatment options.
The table below summarizes
the results of our Phase 2 clinical trial in adults in terms of adverse events.
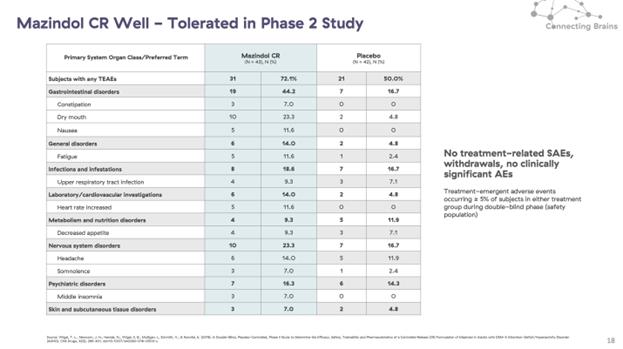
Our Phase 2 clinical trial
evaluated the efficacy, safety and tolerability of Nolazol in a randomized, double-blind, placebo-controlled, multi-center, parallel trial
in 85 adults with a diagnosis of ADHD. Subjects were administered Nolazol or matching placebo once a day for six weeks, dosed flexibly
(between 1mg/day and 3mg/day) during a three-week double-blind optimization period, followed by a three-week double-blind fixed dosing
period.
Efficacy Results
The primary efficacy endpoint
was the change from baseline in the total ADHD-RS score at Week 6, as compared to placebo, and measured by the clinician-administered
ADHD Rating Scale with DSM-5 symptoms, or the ADHD-RS-5. ADHD-RS-5 is a standardized, “gold standard endpoint” validated test
for measuring severity of ADHD symptoms and assessing response to treatment. The scale is based on the ADHD diagnostic criteria as defined
in the DSM-5.
The primary endpoint was met,
showing statistically significant improvement versus placebo, in favor of Nolazol. The LS scores from baseline were -18.9 for Nolazol
and -5.7 for placebo, with a LS mean difference between Nolazol and placebo of -13.2 (95% CI, -18.7, -7.6). The results were found to
be consistent across all sensitivity analyses, establishing that the large placebo-adjusted effect size of 1.09 was not biased by any
of the statistical methods used.
In addition, Nolazol provided
a significant reduction in ADHD-RS-5 scores starting as early as Week 1, which was also observed throughout the full treatment period.
After six weeks of treatment, patients treated with Nolazol demonstrated more than three times the improvement in the ADHD-RS-5 total
score symptoms as compared to placebo treated patients. The figures below summarize the results of the primary endpoint observed in our
Phase 2 trial.
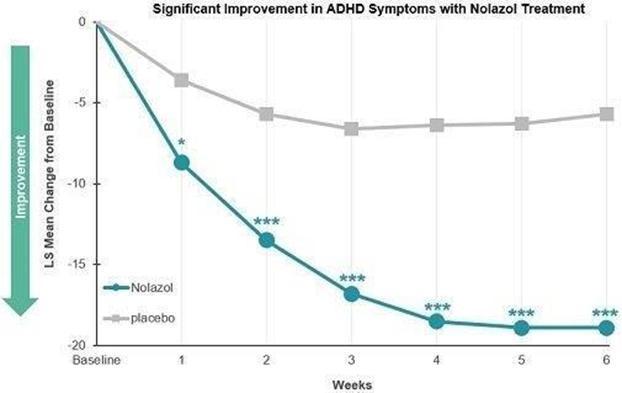
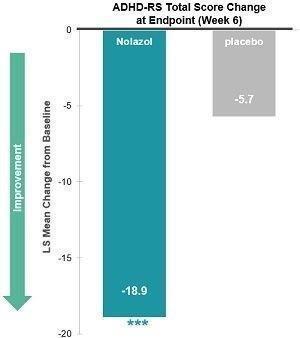
A p-value is a conventional
statistical method for measuring the statistical significance of experimental results. A p-value of less than 0.05 is generally considered
to represent statistical significance, meaning that there is a less than 5% likelihood that the observed results occurred by chance. In
the figure above and all subsequent figures where p-values are included, a p-value of less than 0.05 is represented by “*.”
P-values of less than 0.01 or less than 0.001 are represented by “**” or “***” respectively, and are considered
to have higher statistical significance.
Secondary efficacy endpoints
included patient response to treatment, as measured by the reduction of the ADHD-RS-5 score by at least 30% and by at least 50% from baseline,
and as measured by the Clinical Global Impressions-Improvement, or CGI-I. The CGI-I is a standardized and validated assessment used by
the clinician to rate the severity of a patient’s illness and improvement over time. There was a significant improvement in ADHD-RS-DSM5
scores by Week 6 for Nolazol compared with the placebo. At Week 6, 70% of the Nolazol-treated patients, compared with 21% of the placebo-treated
patients, had at least a 30% reduction in their ADHD-RS-5 score (p < 0.001), and 55% of the Nolazol-treated patients, compared with
15.8% of the placebo-treated patients, had at least a 50% reduction in their ADHD-RS-5 score (p = 0.002). There were significantly more
“responders,” defined as a ≥ 30% reduction from baseline in ADHD-RS-5 scores, present in those receiving
Nolazol compared with placebo at the first assessment point Week 1, and at each subsequent assessment. Furthermore, there were significantly
more “excellent” responders, defined as a ≥ 50% reduction from baseline in ADHD-RS-5 score, compared with
placebo present by Week 2 and at each subsequent assessment point. The excellent response by Week 2 was also evident in the CGI-I analysis,
which indicated significantly more CGI-I responders on Nolazol compared with placebo on Week 2 and at each subsequent visit (p ≤ 0.003).
The sensitivity analyses resulted in a similar magnitude of difference between Nolazol and placebo for all responder definitions. The
figure below summarizes the patient responder results.
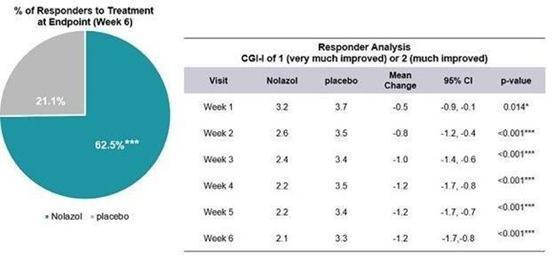
Safety and Tolerability
Nolazol was well-tolerated
and there were no deaths or serious adverse events reported and no discontinuations in the Nolazol treated group due to adverse events
or lack of efficacy. Adverse events reported were mild to moderate and the more prevalent reported events in the Nolazol treated group,
as compared to placebo, included constipation, dry mouth, nausea, fatigue, somnolence, middle insomnia and heart rate increased. Relative
to placebo, on Week 6, the Nolazol treated group had a minimal increase in diastolic and systolic blood pressure, a small increase in
heart rate, and no significant changes in electrocardiography, or ECG, parameters. There were no remarkable findings on physical examination,
hematology, serum chemistry, or urinalysis values from baseline to Week 6 between Nolazol and placebo groups. Mean weight loss of the
Nolazol group was 1.73 kg, compared to a mean weight increase for the placebo group of 1.07 kg.
Phase 1 Clinical Trial
We completed a Phase 1
randomized, open-label, cross-over clinical trial in the United States in 2016, which characterized the pharmacokinetics and
evaluated the safety and tolerability of Nolazol following a single dose in the fasted and fed state in normal healthy adult
subjects. Subjects received a single, fixed dose of Nolazol under fasted conditions in one dosing period and the same single, fixed
dose 30 minutes prior to a high-fat breakfast in another dosing period. Nolazol demonstrated a predictable, dose-dependent, and
linear pharmacokinetics profile. This study is intended to serve as the basis of our Phase 3 clinical trials, if any, by providing a
preliminary understanding of the concentration-time profile of Nolazol under the fed and fasted conditions.
Food administered 30 minutes
after administration of Nolazol resulted in similar exposure and a slightly lower peak concentration compared to administration in the
fasted state, while the time to reach the peak concentration was similar in the fasted and fed states. These results suggest that our
Phase 3 trials will not need to include dosing restrictions in terms of meal timing relative to dose administration, which is an important
feature for increased compliance in ADHD patients. A definitive food-effect study with the final formulation of Nolazol will be conducted
during the Phase 3 program. The figure below shows the pharmacokinetic curve of Nolazol under the fed and fasted conditions utilized in
this clinical trial.
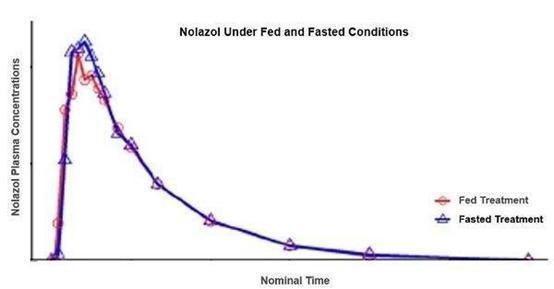
Nolazol was well-tolerated
and there were no deaths, serious adverse events or withdrawals due to adverse events. Adverse events were reported in 30% of the Nolazol-treated
subjects and included dizziness and somnolence and there were no clinically significant changes in laboratory values, ECG, blood pressure,
or heart rate.
Phase 2 Pediatric Clinical Trial
A Phase 2 open-label pilot
study in France was conducted in 2015 by Eric Konofal, one of our founders and Chief Scientific Officer, and others while at AP-HP. This
study evaluated the efficacy, safety, and tolerability of mazindol in 24 children, aged 9 - 12 years, and diagnosed with ADHD. All enrolled
patents had a low rate of response to methylphenidate, which is a first-line treatment in children with ADHD. All patients received the
same fixed dose of mazindol daily for seven days, followed by a three-week drug-free safety observation period. The mean change from baseline
in the parent rated and clinician rated ADHD-RS-IV total score after 7 days of treatment was -24.1, with >90% improvement in ADHD symptoms
from baseline (p<0.0001), pointing to a viable long-acting treatment option, assuming it is shown to be safe, as determined by applicable
regulatory agencies. Additionally, the mean change in the parent rated and clinician rated ADHD-RS-IV total score from end of treatment
(Week 1) to the final observation visit (Week 4) demonstrated statistical significance (p<0.0001), indicating significant alteration
in the level of symptomology of ADHD after mazindol withdrawal. The figure below summarizes the results of the primary endpoint.
Mazindol was well tolerated
in children with ADHD. Adverse events included decreased appetite, headache and abdominal pain and there were no clinically significant
changes in laboratory values, ECG, blood pressure, heart rate, or body weight. This clinical trial provided proof-of concept data, potential
benefit, and supported the advancement into a more expansive Phase 2 trial.
Phase 3 Development Strategy
Having successfully met both
the primary and secondary endpoints in our initial Phase 2 trial, we may plan to further the development of Nolazol as a follow-on or
back-up candidate to support filing for marketing and commercialization approval initially in adults in the United States, followed by
children and adolescents.
Our first Phase 3 clinical
trial may aim to evaluate doses of Nolazol in approximately 260 adults with ADHD, with subjects randomized to receive Nolazol or placebo
for 6 weeks. The primary endpoint being the change from baseline in the ADHD-RS-5 score, which was the primary endpoint in our Phase 2
trial. Our second Phase 3 clinical trial may target to evaluate doses of Nolazol in children and adolescents, with an embedded placebo-controlled
sub-study in a laboratory classroom setting for the children age group. A laboratory classroom study provides a simulation of a real academic
environment, including the potential for interaction and distraction among children, and allows for assessment by trained observers over
the course of a typical extended school day.
Our Research Pipeline
In addition to our product
candidates, Quilience and Nolazol, we have early and mid-stage compounds that we may seek to further develop in the future. We may seek
to develop these other compounds, comprising of NCEs, as well as repurposed compounds, in order to build a pipeline of product candidates
at various stages of development that further complement our rare hypersomnia and complex neurodevelopmental disorder franchises. Additionally,
we intend to continue to invest in our discovery research and development programs, with the goal of adding what we believe to be promising
new compounds and indications to our product candidate development pipeline.
NLS-4
NLS-4 is a next-generation
selective dopamine reuptake inhibitor. We believe that NLS-4 is the only wake-promoting compound without any rebound hypersomnia. This
“hypnolytic effect” of NLS-4 is thought to be due to the compound’s ability to prevent increased sleep need following
sleep loss as supported by the recently published pre-clinical results. With the promising results from the cutting-edge preclinical fatigue
study in animals, and the apparent absence of CYP450 enzyme induction, NLS-4 appears to have a superior profile compared to the widely
used drug modafinil, and potentially represents an important milestone in the development of this next generation wake-promoting agent.
On June 1, 2023, we presented
final results from a preclinical study for NLS-4 at annual meeting of the American Society of Clinical Psychopharmacology (ASCP) in Miami.
The aim of the study was to examine the effectiveness of NLS-4 in comparison with modafinil. NLS-4 is designed to be a more potent next
generation modafinil that does not induce the hepatic toxicity associated with long-term modafinil use. In the study’s Long-COVID
animal model, NLS-4 improved circadian rhythm dysregulation and CFS in subject animals. Based on the results, we believe that NLS-4 should
improve recovery from CFS in humans at a dose that is four times lower than that used for modafinil.
Based on the results, we believe
that NLS-4 offers promise to become a foundational treatment for the chronic fatigue associated with the symptoms of Long-COVID (also
known as Myalgic encephalomyelitis/chronic fatigue syndrome, or ME/CFS). We intend to advance the clinical development of NLS-4, as the
unmet medical need for improved fatigue treatments is growing with more patients surviving infection with COVID-19 and its variants.
ME/CFS is a debilitating chronic
disease with a worldwide prevalence of 0.3–0.8% in the human population. Profound mental and physical fatigue and cognitive
impairment are amongst the key symptoms of ME/CFS. ME/CFS is classified by the World Health Organization as a neurological disease. In
a recent study published in the journal, Clinical Infectious Diseases, a team of researchers examined the risk factors and prevalence
and impact of Long-COVID, among a representative sample of adults in the U.S. and reported that close to 19 million U.S. adults suffer
from Long-COVID. According to a 2023 study by Nature Reviews Microbiology, at least 65 million individuals worldwide are
estimated to have Long-COVID, with cases increasing daily.
Additional Pre-Clinical Compounds
NLS-3
NLS-3 (Levophacetoperane SR)
is a repurposed reverse ester of methylphenidate, a well-documented psychostimulant marketed for treatment of ADHD since the end of the
1950s. Animal experiments and tox studies have demonstrated a very satisfactory safety benefit and more recently, using the behavioral
sensitization test in C57BL/6 mice, it has been reported that NLS-3 (3 mg/kg) vs methylphenidate (6 mg/kg) or d-amphetamine (2 mg/kg)
was not potentially addictive on this contextual sensitization and cross-sensitization to d-amphetamine test pointing-out this stimulant
as less addictive than marketed Scheduled II drugs in ADHD.
NLS-8
NLS-8 (Melafenoxate) is a
melatonin ML1A receptor agonist, improved scopolamine-induced amnesia. In a preclinical study to test the effects of NLS-8 on memory in
a model of Alzheimer’s Disease, the scopolamine-induced amnesia in the novel object recognition test in mice. C57BL/6 male mice,
6 groups (N=16 mice/group), were subjected to two 12-min trials, 90-min apart, in the NOR test: a sample (acquisition) trial during which
they were exposed to two identical objects, and a choice (retention) trial during which they were exposed to two different objects presented,
the familiar object (presented at the sample trial), and the novel object. They received 30 min before the sample trial i.p. injections
of vehicle (control group), of scopolamine, of scopolamine + donepezil (1 mg/kg) or of scopolamine + NLS-8 (50, 100 or 150 mg/kg). Scopolamine
was injected at 1.2 mg/kg. The results of this pre-clinical study showed that NLS-8 improved amnesia induced by scopolamine suggesting
that NLS-8 may improve memory and reduce cognitive symptoms of AD. These effects were significant at doses of 50 and 150 mg/kg and was
close to significant at the dose of 100 mg/kg.
NLS-11
NLS-11 (Benedin) (formerly
SCH-5472) is a norepinephrine and dopamine reuptake inhibitor and muscarinic M1, M2, M3 receptor antagonist. In
a preclinical study, 57BL/6 male mice, 8 groups (N = 16 mice/group), were subjected to the effect of NLS-11 (0.1, 0.5, 1 mg/kg) and compared
to those of vehicle and of donepezil (2 mg/kg). Long-term episodic memory was tested in the NOR test, using a 3-days interval between
the acquisition session (called sample trial) and the retention session (called choice trial). The results of the study showed NLS-11
improved the recognition of the familiar object, i.e. improved memory and suggest that NLS-11 induced a long-term memory improvement which
was significant and of the same extent to that induced by donepezil. At doses tested, NLS-11 did not reduce exploratory behavior, contrary
to donepezil, suggesting that NLS-11 may induce less side effect than donepezil.
NLS-12
NLS-12 (Oxafuramine) is a
norepinephrine and dopamine reuptake inhibitor and muscarinic M4 receptor antagonist, improved long-term episodic memory in
mice. Donepezil was used as positive control drug. A preclinical study was conducted to observe the effects of NLS-12 on the memory in
the novel object recognition test in mice. The dose-ranging study using C57BL/6 male mice: 8 groups (N = 16 mice/group) were subjected
to the effect of NLS-12 (1, 4, 8 mg/kg) and compared to those of vehicle and of donepezil (2 mg/kg). Long-term episodic memory was tested
in the NOR test, using a 3-days interval between the acquisition session (called sample trial) and the retention session (called choice
trial). NLS-12 dose-dependently improved the recognition of the familiar object, i.e. improved memory. This effect was significant at
8 mg/kg and was not significantly different to that of donepezil. The recognition of the familiar object was not modified by NLS-12 (1
mg/kg) and was improved in a non-significant manner by NLS-12 (4 mg/kg). The results of this pre-clinical study suggest that NLS-12 induced
a long-term memory improvement which was significant and of the same extent to that induced by donepezil.
Aexon CNS Platform
Aexon conducts research on
new compounds to address unmet needs in neurodegenerative disorders, defined by the breakdown of neurons over time. Alzheimer’s,
Parkinson’s, Huntington’s, Narcolepsy and Amyotrophic Lateral Sclerosis are just a few examples of brain disorders that have
no cure. Eric Konofal, MD, PhD, who works under a part-time consulting agreement for us as our Chief Scientific Officer, is the president
and founder of Aexon, and owns 59% of Aexon. Alexander Zwyer, our Chief Executive Officer, owns 35% of Aexon. Mr. Zwyer holds no board
or executive position at Aexon.
On March 20, 2024, we
announced that we entered into an exclusive worldwide license agreement with Aexon, a privately held Delaware corporation, under
which we acquired full global development and commercialization rights to Aexon’s Dual Orexin Receptor Agonists platform, new
molecular entities, highly selective dual oral orexin-1 and orexin-2 receptor agonists (OX1R and OX2R) with potential applications
in the treatment of narcolepsy and idiopathic hypersomnia, as well as neuro-degenerative disorders such as Parkinson’s and
Alzheimer’s disease. This license agreement represents a potentially leading next-generation, first-in-class, oral, dual
orexin receptor agonist platform that is expected to address high unmet medical needs and has shown promising results in
pre-clinical in vitro assays. NLSP has plans to initiate proof-of-concept preclinical development following the Merger, subject to
sufficient funding.
The possibility to acquire
this novel and unique platform, consisting of over 300 compounds, bridges the present to the future treatment of sleep disorders as well
as other neuro-degenerative disorders. Orexin receptor pathways play vital regulatory roles in many physiological processes and studies
have shown that orexin receptor pathways are involved in pathological processes of neurological diseases such as obesity, narcolepsy,
depression, ischemic stroke, drug addiction and Alzheimer’s disease. These new compounds, in addition to our current pipeline, including
Mazindol ER for the treatment of narcolepsy, NLS-4 focused on idiopathic hypersomnia, long-COVID and chronic fatigue syndrome, and NLS-11,
addressing Kleine-Levin Syndrome and neurodegenerative diseases, will further complement and strengthen our sleep franchise. NLS will
be uniquely positioned to hold the key to unlock the challenges associated with rare sleep disorders now and in the future.
In addition, we initiated
a comprehensive exploration of new opportunities that align with our core values and strengths, with the goal of diversifying our revenue
streams, mitigating risks and creating lasting value for our stakeholders. As part of this process, the Company is considering a wide
range of options with a focus on maximizing shareholder value, including strategic partnerships, out licensing assets of the Company,
and other future strategic actions. Several strategic partnerships discussions are currently under way and in advanced stages of negotiations.
As always, we continue to approach the sleep medicine market from a holistic perspective. According to Global Narcolepsy Drugs Market
(By Therapeutic Type, Disease Type, End User, Regional Analysis), Pipeline Analysis, Key Company Profiles and Recent Developments - Forecast
to 2030” the global sleep medicine market is estimated to increase to more than $100 billion by 2030, with narcolepsy accounting
for over $6 billion by 2030.
License Agreements
Pegasus Advanced Research SAS License Agreement
In February 2015, Pegasus
Advanced Research, then named NeuroLife-Sciences SAS, or Pegasus, a company owned by certain of our founders, who are also shareholders,
including Eric Konofal, Eric-Jean Desbois, Bruno Figadere and our Chief Executive Officer, Alexander Zwyer, or the Pegasus Founders, entered
into a license agreement with AP-HP, or the AP-HP License Agreement, to obtain a worldwide, sub-licensable license, covering four different
compounds, which included the use of mazindol for the treatment of ADHD, or the Licensed Compounds. As part of the AP-HP License Agreement,
Pegasus was given an option to purchase the Licensed Compounds. In February 2016, after the AP-HP License Agreement was assigned and transferred
to NLS-1, pursuant to an Assignment and Transfer Agreement, or the ATA, NLS-1 purchased the Licensed Compounds from AP-HP for an aggregate
consideration of approximately 2.65 million euros, including reimbursement of certain expenses. On April 1, 2017 and September 20, 2019,
the parties entered into subsequent amendments of the ATA, or the First Amendment to the ATA and the Second Amendment to the ATA, respectively.
Under the Second Amendment to the ATA, NLS agreed to pay Pegasus a royalty of 1.8% of annual net sales (including sublicensee sales) realized
upon the commercialization of products developed on the basis of the Licensed Compounds during the terms of their respective patents;
provided, however, that under certain circumstances, the rate of the royalty payment will decrease. For instance, if a competing generic
product using mazindol for the treatment of ADHD were to become available during the term of the patents covering the Licensed Compounds,
there would be no royalties paid to Pegasus.
Exclusive License Agreement with Eurofarma
In February 2019, we
entered into a License Agreement with Eurofarma Laboratorios S.A., or Eurofarma and the EF License Agreement, which provided
Eurofarma with an exclusive, fee-bearing, non-transferrable (i) distribution right to distribute Nolazol in Latin America and an
(ii) exclusive, fee-bearing, non-transferrable license to our patents and trademarks in connection with the commercialization, if
any, of Nolazol in Latin America. The EF License Agreement is in effect until the later of either (i) ten years from the date of its
execution, or until February 2029, or (ii) until the expiration of the last valid patent relating to Nolazol, subject to early
termination under certain circumstances. Pursuant to the terms of the EF License Agreement, we are responsible for obtaining
regulatory approval to market and commercialize Nolazol in the United States and Eurofarma was responsible for obtaining regulatory
approval in South America; provided, however, that Eurofarma would inform us of any additional information that regulators in Latin
America may require in order to seek marketing authorization which otherwise may not be required by the FDA, or the Supplemental
U.S. Data.
Upon the execution of the
EF License Agreement, Eurofarma paid us $2.5 million. In accordance with the EF License Agreement, we were also entitled to receive milestone
payments as well as royalties from Eurofarma. The EF License Agreement was terminated on August 28, 2024, effective as of September 30,
2024. It was mutually agreed that neither party has any claims against the other in relation to the Agreement or its termination. Consequently,
the deferred revenues amounting to $2,500,000 was realized as of the termination date.
License Agreement with Novartis
On March 10, 2021, we entered
into a License Agreement with Novartis Pharma AG or Novartis, whereby we obtained, on an exclusive basis in the U.S., all of the available
data referred to and included in the original NDA for Sanorex® (mazindol) submitted to the FDA in February 1972. The agreement encompasses
all preclinical and clinical studies, data used for manufacturing including stability and other chemistry manufacturing and controls data,
formulation data and know-how for all products containing mazindol as an active substance, and all post-marketing clinical studies and
periodic safety reports from 1973 onwards. Under the Agreement, we have obtained the same rights on a non-exclusive basis in all territories
outside of the U.S, except for Japan, with the right to cross-reference the Sanorex NDA with non-U.S. regulatory agencies in the licensed
territories. The Agreement includes the right to sublicense or assign the license to third parties, subject to such third parties meeting
certain obligations. As consideration for the license, we agreed to pay Novartis $250,000 upon the signing of the agreement with milestone
payments due as follows: (i) $750,000 payable following the end of a Phase II meeting with the FDA and subject to the FDA’s review
and feedback on the preclinical data from Novartis, with the amount to be reduced to $375,000 in case certain toxicology studies must
be repeated; (ii) $2 million following the earlier of FDA marketing authorization of Quilience or Nolazol; (iii) 1% of any upfront and
milestone payments, if any, from any sublicensees and (iv) $3 million as a one-time payment upon our product candidate reaching $250 million
in cumulative sales.
Intellectual Property
We have developed a
robust patent portfolio of over 100 patents in over 14 countries including the United States, Canada, Australia, China and Japan,
and other European and Latin American countries. Our patent portfolio for Quilience and Nolazol currently includes issued patents in
the United States and Europe covering the use of mazindol for treatment of ADHD and patent applications filed in major countries to
protect our proprietary controlled release formulation (Notice of Allowance recently received for Europe and Canada) for treatment
of ADHD and narcolepsy. Mazindol ER has been grated a formulation patent in the United States, European Union and other countries,
providing protection through March 2037. One of our patents in the United States covering the use of mazindol for the treatment of
ADHD received patent term adjustment, thereby extending the patent term of such patent to August 2028. Additionally, we received
patent approval in Hong Kong covering the use of mazindol for the treatment of ADHD. This patent is expected to expire no earlier
than 2037. We have successfully launched NPP and secured additional patent grants, that coupled with successfully entering into
other expedited development programs, could allow Quilience to reach the market and patients sooner. This further supports our
global strategy with key patents granted the following major markets: Hong Kong, Japan, South Korea, the U.S., Europe & Canada.
Such patents pertain to technology, mechanism of action and application for: (i) sleep disorders, including narcolepsy, idiopathic
hypersomnia and Kleine-Levin syndrome, (ii) ADHD, (iii) neurodegenerative diseases including Lewy body disease, Parkinson’s
and Alzheimer’s, and (iv) treatment of CNS indications associated with sleep disorders.
The following table provides
a description of our key patents and patent applications and is not intended to represent an assessment of claims, limitations or scope
of those patents listed. In some cases, a jurisdiction is listed as both pending and granted for a single patent family. This is due to
pending continuation or divisional applications of the granted case.
| Patent Name and Application Number |
|
Pending
Jurisdictions |
|
Granted/Allowed
Jurisdictions |
|
Expiry Date |
|
Type |
Mazindol combination in the treatment of ADHD
PCT/EP2007/053512 |
|
United States
China |
|
France
United States
Europe
(AT, BE, BG, CH, DE, DK, ES, FI, FR, GB, GR, IS, IT, NL, PL, PT, RO, SE)
Europe (divisional)
(AT, BE, CH, DE, ES, FR, GB, IT, NL, PL, SE)
Canada
Australia
Israel
New Zealand
Morocco |
|
April 11, 2027
(August 22, 2028 for a U.S. patent receiving patent term adjustment of 499 days) |
|
Method of use, composition (EP) |
| |
|
|
|
|
|
|
|
|
Lauflumide and the enantiomers thereof, method for preparing same and therapeutic uses thereof
PCT/EP2012/050881 |
|
|
|
France
United States
Europe
(BE, CH, DE, ES, FR, GB, IT, NL)
Canada
Israel |
|
January 20, 2032 |
|
Method of use (United States), compound
(ex-U.S.) |
| |
|
|
|
|
|
|
|
|
Phacetoperane for the treatment of ADHD
PCT/FR2012/052749 |
|
United States |
|
France
United States
Europe
(AT, BE, CH, DE, DK, ES, FI, FR, GB, GR, IE, IT, NL, PT, TR)
Japan
China
Israel
Canada |
|
November 29, 2032 |
|
Method of use |
| |
|
|
|
|
|
|
|
|
A mazindol ir/sr multilayer tablet and its use for the treatment of ADHD*
PCT/IB2017/000352
*includes narcolepsy and IH |
|
United States
Europe
Australia
Brazil
China
Israel
Japan
New Zealand
Hong Kong |
|
United States
Europe
(AT, BE, CH, DE, ES, FR, GB, IE, IT, LU, MC, NL, PT, PL, SE)
Canada
South Korea
Mexico
Japan |
|
March 8, 2037
(July 16, 2037 for a U.S. patent receiving patent term adjustment of 130 days) |
|
Formulation |
| Patent Name and Application Number |
|
Pending
Jurisdictions |
|
Granted/Allowed
Jurisdictions |
|
Expiry Date |
|
Type |
| Mazindol treatment for heroin dependence and substance use disorder PCT/IB2018/001138 |
|
United States
Europe
Canada
Brazil
China
Japan
South Korea
Mexico |
|
|
|
September 6, 2038 |
|
Method of use |
| |
|
|
|
|
|
|
|
|
|
Use of iron for treating attention deficit hyperactivity
disorder in children PCT/FR2004/001351 |
|
|
|
United States
Europe
(CH, DE, ES, FR, GB, IT, SE)
Canada |
|
June 1, 2024 (December 8, 2029 for a U.S. patent receiving patent term adjustment of 2016 days) |
|
Method of use |
| |
|
|
|
|
|
|
|
|
|
Oxafuramine,
(1R)-N-ethyl-1-[(2R)-oxolan-2-yl]-2-phenylethanamine, hydrochloride and derivatives thereof for treating neurodegenerative diseases
with Lewy Body Disease and/or Alzheimer’s Disease
EP 21305945.4 |
|
Europe |
|
Not Applicable |
|
|
|
Compound for use |
| |
|
|
|
|
|
|
|
|
|
Melafenoxate, 2-(1-adamantylamino)ethyl 2-(4-chlorophenoxy)acetate
and derivatives thereof for treating circadian rhythm sleep disorders with or without neurodegenerative diseases
EP 21305942.1 |
|
Europe |
|
Not Applicable |
|
|
|
Compound for use |
| |
|
|
|
|
|
|
|
|
|
Benedin, Piperidine, 2-benzhydryl-3-hydroxy-N-methyl-, hydrochloride
and derivatives thereof for treating neurological diseases associated with sleep disorders, Central Disorders of Hypersomnolence preferably
Kleine-Levin Syndrome
EP 21305946.2 |
|
Europe |
|
Not Applicable |
|
|
|
Compound for use |
| |
|
|
|
|
|
|
|
|
|
Lauflumide and derivatives thereof for treating chronic fatigue
syndrome and myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS)
EP 21305944.7 |
|
Europe |
|
Not Applicable |
|
|
|
Compound for use |
In addition to our patents
and patent applications, we also rely on unpatented trade secrets, know-how, and continuing technological innovation to develop and maintain
our competitive position. We seek to protect our ownership of know-how and trade secrets through an active program of legal mechanism
including invention assignments, confidentiality agreements, material transfer agreements, research collaborations and licenses to protect
our product candidates. For a more comprehensive discussion of the risks related to our intellectual property, please see Item 3.D. “Risk
Factors - Risks Related to Our Intellectual Property.”
Competition
The biotechnology and pharmaceutical
industries are characterized by rapidly advancing technologies and intense competition. While we believe that our knowledge, experience
and scientific resources provide us with competitive advantages, we face potential competition from many different sources, including
major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public
and private research institutions, which may in the future develop products to treat those diseases that we currently or, in the future,
seek to treat. Any product candidates that we successfully develop and commercialize may compete with existing therapies and new therapies
that may become available in the future.
Many of our competitors have
far greater marketing and research capabilities than us. All of these companies and institutions may have product candidates in development
that are or may become superior to Quilience and Nolazol. Our commercial opportunity would be reduced significantly if our competitors
develop and commercialize products that are safer, more effective, more convenient, have fewer side effects or are less expensive than
either or both of Quilience or Nolazol. Public announcements regarding the development of competing drugs could adversely affect the commercial
potential of either or both of Quilience and Nolazol.
Narcolepsy
We face competition from established
pharmaceutical and biotechnology companies that currently market products for the treatment of symptoms in narcolepsy. There is no cure
and many patients report that their medicines do not improve their complete range of symptoms. For the treatment of both EDS and cataplexy,
we believe that currently our only competitors are Jazz Pharmaceuticals (Xyrem/Xywav®, sodium oxybate) and Harmony Biosciences
(Wakix®, pitolisant). Although only indicated for EDS, our competitors also include Novartis (Ritalin®),
Teva (Provigil®/Nuvigil®, Modafinil/Armodafinil, Axsome (Sunosi®, solriamfetol), Avadel (Lumryz®,
once nightly sodium oxybate), as well as amphetamines, such as Adderall® and Dexedrine®. Other development
stage compounds currently under development include TAK-861 (Takeda’s selective OXR2-agonist), AXS-12 (Axsome, reboxetine) and other
early-stage OX2R agonist (Orexia, Alkermes, Jazz/Sumitomo, Harmony).
The below table highlights
the limitations of certain drug products approved for use in the United States by the FDA for treatment of EDS or cataplexy.
| Product |
|
Cataplexy Approval |
|
Risk of
Abuse
Diversion |
|
|
Limitations |
|
Amphetamines
(Adderall, Dexedrine) |
|
No |
|
High: CII |
|
● |
“Grandfathered” approval only (pre-1938 indication) for Immediate Release (IR) |
|
● |
Tolerance and rebound hypersomnolence |
|
| |
|
|
|
|
|
|
|
|
● |
Short-acting |
|
| |
|
|
|
|
|
|
|
|
|
|
|
Methylphenidate
(Ritalin) |
|
No |
|
High: CII |
|
● |
“Grandfathered” approval only (pre-1938 drug) for IR |
|
● |
Tolerance and rebound hypersomnolence |
|
| |
|
|
|
|
|
|
|
|
● |
Short-acting |
|
| |
|
|
|
|
|
|
|
|
|
|
|
Solriamfetol
(Sunosi) |
|
No |
|
Low: CIV |
|
● |
Efficacy for controlling cataplexy not available |
|
● |
Risk for drug induced high-blood pressure that can go undetected |
|
| |
|
|
|
|
|
|
|
|
|
|
|
Modafinil /Armodafinil
(Provigil/Nuvigil) |
|
No |
|
Low: CIV |
|
● |
Potential for serious rash, including Stevens-Johnson syndrome |
|
● |
Substrate, inducer, and inhibitor of CYP450 isoenzymes, which significantly increases the risks for drug-drug interactions |
|
| |
|
|
|
|
|
● |
Cases of major fetal congenital malformations |
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
Pitolisant
(Wakix,) |
|
Yes |
|
Not determined |
|
● |
Pitolisant may reduce efficacy of oral contraceptives, and so |
|
● |
Unclear efficacy; partially failed Phase 3 program |
|
| |
|
|
|
|
|
|
alternative methods of contraception should be utilized |
|
● |
Data suggests it’s only effective in 1/3 of patients |
|
| |
|
|
|
|
|
|
|
|
|
|
|
Sodium oxybate
(Xyrem/Xywav, Lumryz) |
|
Yes |
|
High: CIII, CI penalties for diversion |
|
● |
Very short acting; requiring two nighttime doses |
|
● |
Can take up to 3+ months for response |
|
| |
|
|
|
|
|
● |
Not effective against EDS, so used in combination with stimulants or modafinil |
|
● |
Known as the “date rape drug” |
|
| |
|
|
|
|
|
● |
Because of the risks of depression, abuse, and misuse, Xyrem is available only through a restricted distribution program called the Xyrem REMS Program |
|
● |
Xyrem is a CIII controlled substance as sodium salt of gamma hydroxybutyrate (GHB), a Schedule I controlled substance. Abuse or misuse of illicit GHB is associated with CNS adverse reactions (acting as a CNS depressant), including seizures, respiratory depression, decreased consciousness, coma, and death |
|
| |
|
|
|
|
|
● |
Potential life-threatening adverse effects |
|
|
|
|
ADHD
We face competition from established
pharmaceutical and biotechnology companies that currently market a range of CII stimulants to treat ADHD. Our primary competitors include
Takeda Pharmaceutical Company Ltd. (Vyvanse®, Adderall® and Mydayis®), Neos Therapeutics
Ltd. (Adzenys® XR-ODT and Contempla® XR-ODT), Eli Lilly & Co. (Strattera®), Novartis
AG (Focalin®) and Janssen Pharmaceutica N.V., a subsidiary of Johnson & Johnson (Concerta®). The below
table provides a more in-depth breakdown of Nolazol against certain competing pharmaceutical products, based on the current scheduling
of mazindol by the DEA.
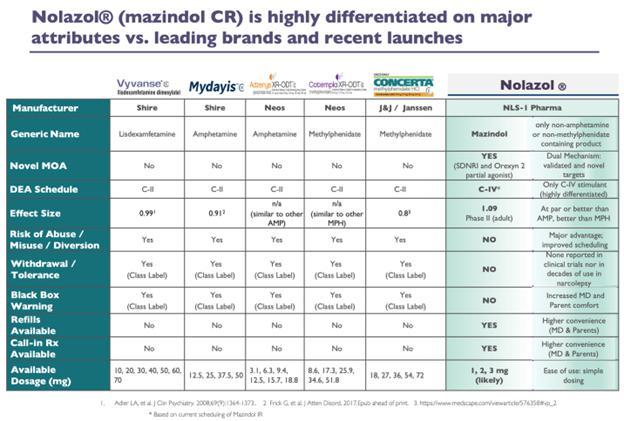
Research and Development Strategy
Subject to obtaining
sufficient funding, we aim to continue conducting research and development activities to expand the commercial potential of both
Quilience and Nolazol, while continuing to examine the development of compounds that could serve as effective treatments for other
CNS disorders. We sponsor and conduct clinical research activities with investigators and institutions to measure key clinical
outcomes that are necessary in order for us to be able to file an NDA with the FDA and equivalent filings with other regulatory
authorities. Our research and development efforts are focused primarily in the following areas and serve as a basis for future
development, if any, of a more diverse product pipeline, of which certain product candidate leads, such as NLS-4 are in preclinical
development stages. As we navigate the competitive landscape of our industry, while focusing on development of our product
candidates, we also intend to continually pursue out-licensing agreements and asset sale transactions that we believe will allow us
to drive greater value for our shareholders. Key elements of our research and development strategy include the following:
| ● | Efficiently
advance our lead product candidate, Quilience, and follow-on product candidate, Nolazol, through marketing approval. We plan
to first advance the development of Quilience, followed by the development of NLS-4 (follow on). If successful, we plan to initially
file for marketing approval in the United States and potentially also in the EU for Quilience and subsequently develop NLS-4 (NCE) for
post-COVID chronic fatigue syndrome for global markets. |
| |
● |
Find partners for out-licensing and asset sale agreements. While we continue with our goal of progressing our product candidates, Quilience and Nolazol, on our own into further clinical development in order to initiate commercialization of such product candidates, we may seek to enter into transactions to sell or out-license Quilience, Nolazol or certain other product candidates or intellectual property that we develop. This strategy allows us to potentially create value for our shareholders ahead of our approval timelines. |
| |
|
|
| |
● |
Reduce clinical and regulatory risk, limit development costs, and accelerate time to market. Our product candidates, Quilience and Nolazol, incorporate a known molecule in a proprietary CR and ER formulation. The former immediate release formulation of mazindol has a well-established safety record from its long history of clinical use across the United States and several countries in Europe and, as a result thereof, a well-characterized safety profile that has allowed us to rapidly begin conducting clinical development of Nolazol and to generate supportive phase 2 data in a study conducted in 85 patients with ADHD in the United States in May 2017 and complete a Phase 2 study in patients with narcolepsy in the United States. We believe that this strategy also allows us to potentially seek FDA approval using the 505(b)(2) regulatory pathway for both product candidates. |
| |
● |
Develop products with differentiated pharmacological profiles. We are developing product candidates with dual mechanisms of action. For example, Quilience and Nolazol utilize a dual mechanism of action, resulting in a unique pharmacological profile targeting multiple neuronal pathways that are widely thought to be disrupted and lead to the disorders targeted by our product candidates. We believe that products with clearly differentiated features, as compared to currently available drug therapies, will be attractive to patients and physicians and will provide us with a competitive commercial advantage. |
| |
● |
Maximize the therapeutic potential of our
existing targets and product candidates. Given the central physiological roles played by the distinct targets of our lead and
follow-on product candidates, we believe that there is significant potential for us to address multiple indications and our goal is to
expand the therapeutic and commercial potential of our existing product candidates to additional indications. For example, we plan to
explore the development of Quilience for the treatment of IH, another rare CNS disorder for which Quilience received orphan drug designation
in the U.S. and in Europe.
|
| |
● |
Deploy our value-driven approach to broaden our sleep-related product portfolio. Our team has extensive experience in CNS research and a strong record of publication in peer-reviewed journals and we plan to develop additional product candidates to treat indications with a high unmet medical need, and may seek to in-license from or collaborate with third parties to develop product candidates that we believe are highly differentiated, promising therapeutic candidates that address major unmet clinical needs. Our scientifically rigorous approach to evaluating new opportunities includes a robust asset evaluation of key factors, including the unmet medical need, biological rationale, safety profile, as determined by applicable regulatory agencies, feasibility of clinical development, potential for accelerated development path, regulatory approval, intellectual property position, competitive landscape and commercial potential. |
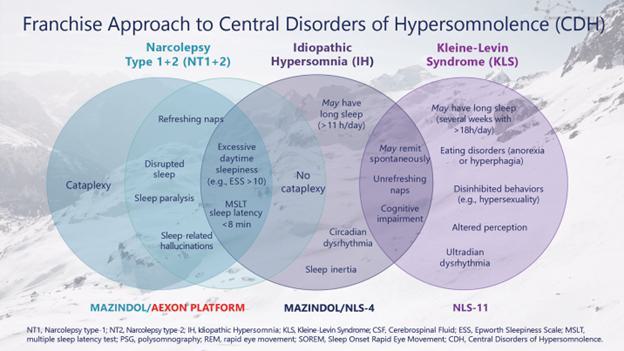
Manufacturing and Suppliers
We do not own or operate manufacturing
or distribution facilities for the production of our product candidates and we currently rely, and expect to continue to rely, on third
parties for the manufacturing, packaging, labelling and distribution of our product candidates for pre-clinical and clinical testing,
as well as for future commercial manufacturing, if our product candidates receive marketing approval. We require all of our contract manufacturing
organizations to conduct manufacturing activities in compliance with cGMP requirements and although we rely on manufacturers, we have
engaged with consultants with significant technical, manufacturing, analytical, quality, regulatory, including cGMP, and project management
experience to oversee our third-party manufacturers. This approach allows us to maintain a more efficient infrastructure while enabling
us to focus our expertise on developing and commercializing our product candidates. Reliance on third-party providers may expose us to
more risk than if we were to manufacture product candidates ourselves.
In December 2019, we
entered into an agreement with Cambrex High Point for the production of our drug substance, pursuant to which, upon completion of
purchase orders, Cambrex High Point may manufacture mazindol for us. We obtain our supply of the finalized drug product from another
third-party manufacturer, CoreRX with whom we signed an agreement in February 2021. We do not currently have any contracts binding
us to use supply or production services provided for under such agreements. We expect to continue to rely on third-party
manufacturers to produce sufficient quantities of our product candidates and their component raw materials for use in our internal
research efforts and clinical trials and in relation to any future commercialization of our product candidates. Our third-party
manufacturers are responsible for obtaining the raw materials necessary to manufacture our product candidates, which we believe are
readily available from more than one source. Additional third-party manufacturers are and will be used to formulate, fill, label,
package and distribute investigational drug products and eventually our products, if and when our product candidates receive
approval. This approach allows us to maintain a more efficient infrastructure while enabling us to focus our expertise on developing
and commercializing our product candidates. We believe that our current supplier and manufacturers have the capacity to support both
clinical supply and commercial-scale production, but we do not have any formal agreements at this time for such supply and
production, and we may also elect to enter into agreements with additional or alternative parties in the future.
Commercialization, Sales and Marketing
Given our stage of development,
we do not currently have an established internal sales, marketing or distribution infrastructure. If our product candidates are approved
for marketing, we intend to commercialize our product candidates either alone or in partnership with others, where appropriate, to maximize
the value of our product candidates. We expect to build our commercial infrastructure using a focused and efficient approach, initially,
establishing market access, sales and marketing capabilities in a targeted manner that is appropriate for the relevant product opportunity.
We believe that this approach will allow us to effectively reach patients and physicians and to maximize the commercial potential of our
product candidates.
Kadimastem Business
Kadimastem is a clinical-stage pharmaceutical
company focused on developing and manufacturing “off-the-shelf”, allogeneic, proprietary cell products based on its technology
platform for the expansion and differentiation of human embryonic stem cells, or hESCs, into functional cells.
Kadimastem’s vision is to:
| ● | Replace, restore and repair the functionality of diseased
and malfunctioning cells in various degenerative diseases by transplantation of healthy and functional cells; and |
| ● | Commercialize its proprietary cell lines optimized for both
the cure of diabetes and the treatment of ALS, and other neurodegenerative diseases. |
Kadimastem is primarily developing two products:
| 1. | AstroRx®, Kadimastem’s lead product,
which is an off-the-shelf cryopreserved cell therapy product in clinical development for the treatment for ALS and in pre-clinical studies
for other neurodegenerative indications. AstroRx® is comprised of fully differentiated astrocytes, which are mainly cells
that support the CNS. |
| 2. | IsletRx is Kadimastem’s treatment for diabetes. IsletRx
is comprised of functional, insulin and glucagon producing and releasing pancreatic islet cells, intended to treat and potentially cure
patients with Type 1 diabetes and possibly Type 2 insulin dependent diabetes. |
Kadimastem was founded by Professor Michel Revel,
Chief Scientific Officer of Kadimastem and Professor Emeritus of Molecular Genetics at the Weizmann Institute of Science. Professor Revel
received the Israel Prize for the invention and development of Rebif®, a multiple sclerosis blockbuster drug sold worldwide.
Kadimastem is currently listed on the TASE (TASE: KDST).
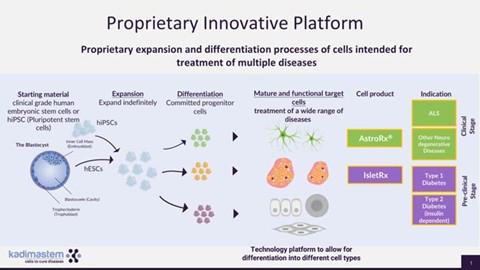
Kadimastem Overview
Kadimastem is a clinical stage biotechnology company,
with a unique platform for cell therapy that enables the production of off-the-shelf cell-based products for the treatment of unmet medical
needs. Kadimastem operates in the field of development of cell therapy, regenerative medicine, for the treatment of, among others, ALS,
an incurable disease, for which Kadimastem received an orphan drug designation status from the FDA, as well as cell therapy in the field
of regenerative medicine for the treatment of diabetes, a disease that affects hundreds of millions of people worldwide, and its product
has the potential to provide a cure for the disease. Kadimastem intends to be a leading company in the field of regenerative medicine
products for the treatment of neurodegenerative diseases and a cure for diabetes.
Kadimastem is developing revolutionary regenerative
therapies based on stem cells-derived therapeutic cells, moving away from the traditional curative therapies. The technology has been
developed as a platform enabling the manufacturing of islet-like endocrine cells and glia restricted progenitors thus having potential
applications for diabetes, and for neurodegenerative diseases such as ALS. The therapy is scalable and industrialized, to be commercialized
as a stable “off the shelf” product and reduce the cost of treatments. For this, Kadimastem uses pluripotent cells (e.g. embryonic
stem cells — hESCs) that have a unique ability to multiply infinitely without losing their “naivety” and to
be able to become any cell type. The cell therapy products manufactured under Good Manufacturing Practices, or GMP, guidelines (similar
to traditional therapeutics) in order to reach optimal clinical results. Kadimastem developed a new process to differentiate the cells
in the lab to their mature phenotype, before their implantation to the patient, unlike other technologies which transplant immature precursor
cells. Thus, Kadimastem believes that its process will markedly enhance the efficiency of the treatment. The transplantation of immature
cells depends on exact signals from the surrounding tissues to support their maturation into functional cells. This process in vivo cannot
be controlled, and as such the outcome of such is variable. In contrast, mature cells are ready to use and do not require specific signals
to confer their function. Furthermore, we have observed, based on our data, that the transplantation of these mature cells enhances treatment.
Furthermore, we have observed, based on our data, that the transplantation of these mature cells slows disease progression. In NCT0348205010
phase 1/2a clinical trial, 10 ALS patients were injected with AstroRx® and as a result, in the first three months post treatment the
disease progression was slowed significantly (study outline is described below beginning on page 129).
Kadimastem implements a technological platform
that uses pluripotent stem cells, or PSCs, either embryonic stem cells and/or induced pluripotent stem cells for the development and production
of various active cells as off-the-shelf products for the treatment of a wide range of diseases. Its technological platform includes processes
for the development, production and biobanks of cells at various stages of differentiation. As of the date of this annual report, Kadimastem
focuses on the development of cell therapy products in the field of regenerative medicine for the treatment of various diseases with an
emphasis on (a) treatment and/or cure of neurodegenerative diseases of the CNS, with the purpose of finding a treatment for ALS,
and (b) development of a cure for Type 1 diabetes and possibly in the future, for Type 2 diabetic patients which are insulin dependent.
Regenerative medicine is an innovative medical
research field that focuses on regeneration of tissue/organs harmed due to disease, injury or due to birth defects in patients, using
one of the following two ways: (1) creating new cells, organ parts or tissues under laboratory conditions, or using donor cells,
organs or parts of organs transplanted into the patient’s body in order to replace the cells or tissues damaged by disease; (2) finding
and developing drugs that will help induce a process of spontaneous regeneration of the damaged tissue/organ by encouraging the adult
stem cells that are regularly present in the tissue, divide, differentiate and take their place in the affected area.
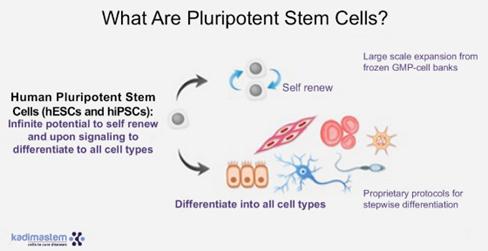
Below is a discussion on Kadimastem’s two
primarily drug products, AstroRx®, for the treatment for ALS, and IsletRx, for the treatment of diabetes.
AstroRx® — Development of a Drug for the Treatment
of Neurodegenerative Diseases (With an Emphasis on ALS)
Development of Cell Therapy in the Field of
Regenerative Medicine for Patients with Neurodegenerative Diseases
Neurodegenerative diseases are degenerative diseases
of the CNS. These diseases are characterized by massive mortality of neurons. The prevention of neurodegeneration represents one
of today’s most significant unmet medical needs. The development of therapies that preserve neuron health present unique challenges,
including an imperfect understanding of underlying biology and a lack of translation of activity observed in preclinical studies to results
in clinical trials. Kadimastem believes that currently approved therapies for many neurodegenerative diseases are generally only symptom
modifying and have demonstrated limited efficacy. Accordingly, Kadimastem believes there remains an urgent need for novel approaches to
address most neurodegenerative diseases, especially for progressive and severe conditions such as ALS.
Currently, there are no FDA approved-treatments
to stop the disease’s progression or prevent onset. There are two FDA-approved treatments which attempt to slow ALS progression;
Riluzole and Radicava. There are a number of ongoing trials examining potential treatments for ALS.
ALS is a multifactorial disease and therapeutic
approaches should consider the multiplicity of pathophysiological mechanisms that underlie motor neuron, or MN, degeneration in this disease.
Though MNs are the main affected cells in the disease, there is involvement of malfunctioning astrocytes in the pathogenesis and progression
of ALS. This notion supports the rationale that transplantation of healthy human astrocytes into the CNS of ALS patients may compensate
for the malfunctioning astrocytes and rescue remaining MNs by supply supportive factors, clues and functions. The product AstroRx®
is composed of healthy human astrocytes derived from hESCs which allows ready scale-up of large numbers of donor cells. AstroRx®
cells are manufactured in compliance with GMP guidelines. Once manufactured, the cells are resuspended in PlasmaLyte A (a physiological
solution) to formulate the AstroRx® product. The product is loaded into a syringe and delivered to the physician at point-of-care
for intrathecal injection, or IT injection, into the CSF of ALS patient following minimally invasive lumbar puncture, or LP.
Kadimastem’s Solution
The cell therapy
Kadimastem is developing to treat ALS, AstroRx®, is based on the transplantation of allogeneic glial cells (mainly
cells that support the CNS, or astrocytes) that have differentiated from pluripotent stem cells, thereby returning to the recipient
the motoneuron-supporting environment. It is assumed that this procedure will moderate the mortality rate of the neurons, and maybe
even stop it, so that the quality of life and life expectancy of the patients will improve. Astrocyte function in neuronal support
is also impaired in other neurodegenerative diseases such as multiple sclerosis, progressive multiple sclerosis, frontotemporal
dementia, or FTD, Alzheimer’s, Parkinson’s and glaucoma.
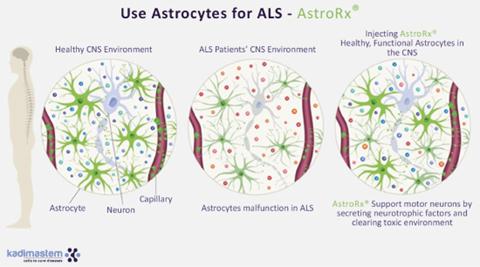
Mechanism of Action: AstroRx®
was demonstrated to promote neuroprotection and to maintain homeostasis through several mechanisms including reuptake of excessive glutamate,
neutralizing oxidative stress, immunomodulation and secretion of a variety of potent neurotrophic factors. For example, disruption of
the astrocytic TNFR1 — GDNF axis accelerates MN degeneration and disease progression, as the levels of the protective
agents for MNs, glial-derived neurotrophic factor, are reduced. AstroRx® were tested in vitro, in vivo (animal studies)
and on human subjects, as described in the following peer reviewed papers:
PMID: 36788520: Safety and efficacy of first-in-man
intrathecal injection of human astrocytes (AstroRx®) in ALS patients: phase I/IIa clinical trial results. This paper describes
the results of Phase 1/2a NCT0348205010 clinical trial (further described on page 137).
PMID: 29871694: Safety and efficacy of human
embryonic stem cell-derived astrocytes following intrathecal transplantation in SOD1G93A and NSG animal models. This
paper describes the development of good manufacturing practice-grade protocol for generation of astrocytes from human embryonic stem cells
(AstroRx®), their in vitro function, their efficacy in ALS animal model (hSOD1G93A transgenic mice) and their
safety and biodistribution in a 9-month study conducted in immunodeficient NSG mice under good laboratory practice conditions. In vitro,
AstroRx® possess the activities of functional healthy astrocytes, including glutamate uptake, promotion of axon outgrowth
and protection of MNs from oxidative stress. A secretome analysis shows that AstroRx® also secretes several inhibitors of
metalloproteases as well as a variety of neuroprotective factors (e.g. TIMP-1, TIMP-2, OPN, MIF and Midkine). Intrathecal injections of
the AstroRx® into transgenic hSOD1G93A mice and rats significantly delayed disease onset and improved motor
performance compared to sham-injected animals. A safety study in immunodeficient mice showed that intrathecal transplantation of AstroRx®
demonstrated positive results. Transplanted AstroRx® attached to the meninges along the neuroaxis and survived for the
entire duration of the study without formation of tumors or teratomas. Cell-injected mice gained similar body weight to the sham-injected
group and did not exhibit clinical signs that could be related to the treatment. No differences from the vehicle control were observed
in hematological parameters or blood chemistry.
PMID: 32848579: Rising Stars: Astrocytes as a Therapeutic Target
for ALS Disease
This paper describes the role of Astrocytes malfunction
in several neurodegenerative diseases and specifically in ALS. Astrocytes impaired biological functionality were implicated in multiple
neurodegenerative diseases, including ALS, FTD, Parkinson’s, and Alzheimer’s. In ALS
disease patients, A1 reactive astrocytes were found to play a key role in the pathology of ALS disease and death of motor neurons, via
loss or gain of function or acquired toxicity. The contribution of astrocytes to the maintenance of motor neurons by diverse mechanisms
makes them a promising therapeutic candidate for the treatment of ALS. Therapeutic approaches targeting at modulating the function of
endogenous astrocytes or replacing lost functionality by transplantation of healthy astrocytes, may contribute to the development of therapies
which might slow down or even halt the progression ALS diseases. The proposed mechanisms by which astrocytes can potentially ameliorate
ALS progression and the status of ALS clinical studies involving astrocytes are discussed.
In the Phase 1/2a trial of
AstroRx®, when applied to humans, these effects were not observed at 6- and 12-month follow-up time points. As so, Kadimastem
believes that it is more likely that AstroRx® astrocytes will be more effective in treating diseases than single-molecule
based drugs, e.g. Riluzole (assumed to improve glutamate uptake and to reduce excitotoxicity), or Edaravone (a reactive oxygen species,
reducing oxidative stress). AstroRx® astrocytes’ activity may include both drugs’ modes of action, in addition
to secretion of multiple neurotrophic factors that act through multiple pathways. Importantly, this astroglial-based approach should
be therapeutic regardless of the etiology of the ALS — familial (independent of genotype) as well as sporadic — because
it attacks final common molecular pathways of MN demise, which contrasts with some present genetic approaches.
Mode of Action: AstroRx®
is based on differentiated clinical grade cells derived from embryonic stem cells it received under licensing agreements and
technologies developed by it to expand pluripotent stem cells and differentiate them into cells supporting the central nervous
system (astrocytes), and then transplant them into patients. In healthy individuals, astrocytes support neuronal health and function
by releasing growth factors, removing toxins, and reducing environmental stressors such as free radicals and excess glutamate or potassium.
Strengths
Kadimastem’s vision is to act to find cell
therapy treatments for incurable diseases that impair patients’ life expectancy and quality of life, such as ALS. The strategy
is to replace the functionality of malfunctioning cells even within some distance from the original tissue/organ that is difficult or
impossible to replace. The activity of malfunctioning astrocytes can be restored using “healthy” astrocytes cells that perform
their physiological role upon their implantation.
Kadimastem believes it has the potential to transform
the lives of individuals living with devastating neurodegenerative diseases beginning with patients suffering from ALS. Kadimastem’s
key competitive strengths include:
| ● | Kadimastem developed scalable manufacturing capabilities and
industrialization capabilities for AstroRx®. |
| ● | Kadimastem has a well differentiated, solid, and diversified
technology platform. |
| ● | AstroRx® can be utilized for other neurodegenerative
indications. Some of the additional indications in the pipeline were already evaluated pre-clinically. |
| ● | Kadimastem has experienced leadership that will continue post-merger. |
| ● | Strong global IP position and differentiation. |
In the field of regenerative medicine,
Kadimastem’s product candidate, AstroRx®, is a cell therapy product that is manufactured from a frozen ampoule
of immature astrocyte cells (production batches). The process of production of the AstroRx® cells takes about one
month, conducted by a manufacturing team in GMP suits. Utilizing a frozen, off-the-shelf product improved the streamlined production
processes and allow Kadimastem to transport AstroRx® frozen cell product from one (or more) production facility to
anywhere in the world, at any point in time, without Kadimastem having to synchronize the production process with the exact date of
injection of the product to any patient. Kadimastem conducted several trials that supported the stability of the frozen
AstroRx® product and that the freezing process did not change the characteristics of the product. In addition,
AstroRx® successfully passed toxicity studies. The final chemistry, manufacturing and controls data was submitted to
the FDA in the Investigational New Drug, or IND, application and the FDA approved the conduction of Phase IIa clinical trial in
March 2023. Kadimastem intends to initiate a Phase IIa multisite study in the United States shortly following the
closing of the Merger.
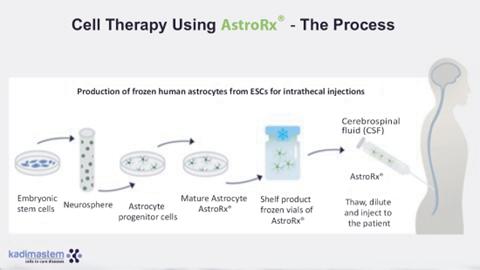
As described in more detail below, in 2021 Kadimastem
completed a Phase 1/2a clinical trial of AstroRx® that began in April 2018, with the first patient treated in November
2018. The study was conducted at Hadassah Medical Center, Israel. Study protocols were approved by the Israeli Ministry of Health and
the institutional review board of Hadassah Medical Center in Jerusalem, Israel. Eligible participants were aged between 18 and 70 years
with a diagnosis of probable or definite ALS by revised El Escorial Criteria, within two years of diagnosis. The ALS Functional Rating
Scale-Revised, or ALSFRS-R score was higher than 30, and slow vital capacity, or SVC, was 70% or more of the predicted normal value for
height, age, and sex. Participants were either not receiving riluzole and/or edaravone or were on a stable dose for more than 30 days.
Potential patients were excluded for the following reasons: past infection or a positive test for hepatitis B, hepatitis C or HIV (human
immunodeficiency virus), need for respiratory support, renal failure, impaired hepatic function, body mass index under 18.5 or 30 or above,
significant cardiac disease, diabetes, autoimmune diseases, chronic severe infection, malignant disease or any other disease or condition
that may risk the patient or interfere with the result of the study. By the end of December 2019, ten patients were treated with
AstroRx®: five patients from Cohort A had been treated with 100 million AstroRx® cells injected into
their spinal fluid. Subsequently, five patients from Cohort B were treated with 250 million AstroRx® cells, with the
last patient being treated in December 2019. During 2020, clinical data was collected for the treated patients for up to 12 months.
Considering the spread of COVID-19 in Israel and following the data and safety monitoring board recommendation to suspend the continuation
of the clinical trial in the third and fourth treatment groups, Kadimastem decided to discontinue the recruitment to additional treatment
groups in ALS Phase 1/2a trial.

Safety outcomes: Nine out of 10 (90%) of treated
patients completed the six-month follow-up, and 6 patients (60%) completed the twelve-month follow-up. All patients reported a total of
86 treatment-emergent adverse events, or TEAE. None of the TEAEs was deemed to be associated with AstroRx® itself. 63 TEAEs
were mild, 19 were moderate, and 4 were severe. Six patients developed a total of 9 serious TEAEs after the treatment, 2 patients in Group
A and 4 patients in Group B. The most frequent TEAE was post lumbar puncture headache, associated with IT injection procedure of the cells,
and reported by 50% of the patients. Additional procedure-related TEAEs included pain in the injection site (30%), arthralgia, back pain,
muscle contraction, and pain in the leg, each reported by 10% of the patients. All procedure-related adverse events, or AEs, were graded
as mild to moderate, and all were resolved. Three patients reported 4 AEs related to mycophenolate mofetil, including headache, nausea,
anemia, and hyperhidrosis. All the immunosuppression-related AEs were graded as mild to moderate, and all were resolved.
The main outcome efficacy measure in the study
was ALSFRS-R. At baseline visit (1 day before treatment) the mean ALSFRS-R score was 35.6 ± 3.7, 34.2 ± 7.0 and
34.9 ± 5.3, for Group A, Group B, and combined Group A + B, respectively. The mean decline in the ALSFRS-R slope for patients in
Group A was − 0.88/month during the run-in (3 – 4 months before treatment). In the first 3 months after
AstroRx® cell injection, the mean decline of the ALSFRS-R slope was attenuated to − 0.3/month (p = 0.039), reflecting
an attenuation of 66% in ALSFRS-R deterioration. Combining the data of both groups demonstrated an attenuation of 53% in ALSFRS-R over
the first 3 months post AstroRx® IT injection (p < 0.001) (a p-value is a conventional statistical method for measuring
the statistical significance of experimental results; p-value of less than 0.05 indicates statistically significant result), which was
not maintained at 6- and 12-month follow-up. The change in the ALSFRS-R slope was also analyzed in a subpopulation of rapid progressors
from both groups (n = 5). Rapid progressors were defined as patients who deteriorated ≥ 1.1 ALSFRS-R points per month during the
run-in period. The mean improvement in ALSFRS-R slope between the run-in period and 3-month follow-up in these patients was 58% (−
1.58/month vs. − 0.65/month, p < 0.001). Also in this subpopulation, after 3 months post single dosing the ALSFRS-R slope
returned to a similar rate that was recorded before treatment (Fig. 2). An improvement ≥ 25% in the ALSFRS-R slope is considered
clinically meaningful. The individual ALSFRS-R slopes demonstrated an improvement of at least 25% in ALSFRS-R slope between the run-in
and 3-month follow-up in 80% of the patients (4 patients in each group, data not shown).
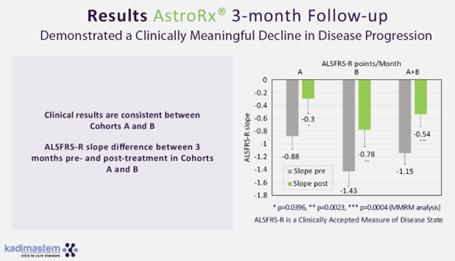
At 6 and 12 months after treatment, the ALSFRS-R
deterioration rate was − 0.76/month and − 0.82/month, respectively, similar to that observed during run-in. The mean deterioration
of ALSFRS-R slope in Group B (− 1.43/month) during the run-in was greater than Group A (− 0.88/month). Similar to Group A,
the ALSFRS-R deterioration rate during the first 3 months after treatment decreased to − 0.78/month (p = 0.002), representing
an attenuation of 45% in ALSFRS-R decline. As observed in Group A, the attenuation of ALSFRS-R decline over the first 3 months post-treatment
was not maintained at 6 and 12 months post-treatment (− 1.59/month and − 1.39/month, respectively).
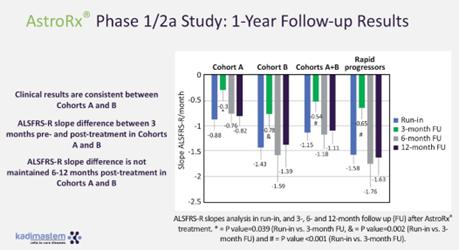
FDA approved planned Phase 2a
study: In March 2023, Kadimastem received approval from the FDA regarding its AstroRx®
multisite-site Phase 2a clinical trial study in the United States based on the IND application. The application includes a detailed
clinical synopsis approved by the FDA. Briefly, the studies titled: “A Prospective, Randomized, Double-Blind, Placebo
Controlled, Phase 2a Study For 6 Months, followed by a 6-Month Open-Label Extension to Evaluate the Safety, Tolerability, And
Effectiveness of Repeated Administration of Cryopreserved AstroRx® in Patients with Amyotrophic Lateral Sclerosis
(ALS)”. This study will be conducted in participants with a sporadic or familial ALS, who meet the El Escorial criteria of
probable, or definite criteria for a diagnosis of ALS, with disease onset of 18 months or less, from first symptoms until screening.
At screening, eligible participants must be between ages 18-80 years old, have an ALSFRS-R score ≥35, and a SVC ≥70% of
predicted capacity for age, height and gender. Participants on a stable dose of riluzole and/or edaravone or PB (sodium
phenylbutyrate)/TURSO (taurursodiol), and those not taking riluzole and/or edaravone or PB/TURSO at screening are eligible for
inclusion as long as they meet specific protocol requirements. Approximately 30 adult participants will be enrolled into the
double-blind study with a randomization ratio of 2:1 (20 participants in the AstroRx® arm and 10 participants in the
placebo arm). Kadimastem began preparations for the clinical trial including extensive contact with key opinion leader and clinical
centers in the United States and signing a tech-transfer agreement with qualified Contract Development and Manufacturing
Organization for the manufacturing of clinical grade AstroRx® product.
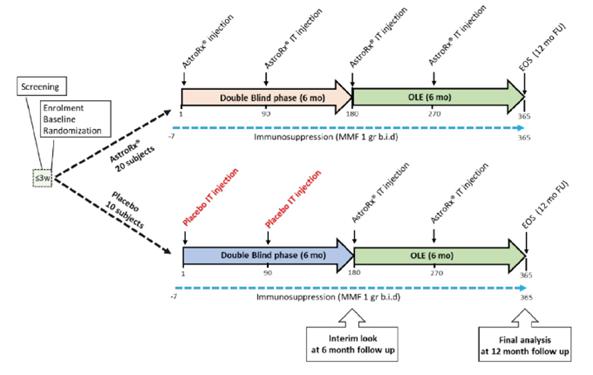
Kadimastem’s AstroRx® has
received orphan drug status from the FDA, a status given to certain drugs called orphan drugs, which show promise in the treatment, prevention,
or diagnosis of orphan diseases. Kadimastem believes that the granting of orphan drug status for the AstroRx® product is
an acknowledgment of the uniqueness and medical potential of the product and will give a significant boost to its entry into the market
once it’s approved for marketing. The FDA grants orphan drug status in order to provide an incentive for the development of drugs
and medical treatments. Companies whose drugs have been awarded this status enjoy exclusive marketing rights for the drugs for seven years
after marketing approval is obtained. This recognition is also useful in obtaining grants and financial concessions in development and
registration of drugs for marketing.
The FDA has authority to grant orphan drug designation
to a drug or biological product to prevent, diagnose or treat a rare disease or condition. Orphan drug designation qualifies sponsors
for incentives including tax credits for qualified clinical trials, exemption from user fees, and potential seven years of market exclusivity
after approval. Orphan drug status provides faster testing tracks and responses from the FDA and other regulatory agencies. Orphan drug
status designation neither shortens the development time nor regulatory review time of a drug nor does it increase the likelihood for
any approval in the regulatory review process. Sponsors requesting designation of the same drug for the same rare disease or condition
as a previously designated product must submit their own data and information to support their designation request. Orphan drug designation
is a separate process from seeking approval or licensing. Drugs for rare diseases go through the same rigorous scientific review process
as any other drug for approval or licensing.
Competition
Kadimastem faced substantial competition in
all fields of business in which Kadimastem engages. That competition is likely to intensify as new products and technologies reach
the market. Superior new products are likely to sell for higher prices and generate higher profit margins if acceptance by the
medical community is achieved. Those companies that are successful at being the first to introduce new products and technologies to
the market may gain significant economic advantages over their competitors in the establishment of a customer base and track record
for the performance of their products and technologies. Such companies will also benefit from revenues from sales that could be used
to strengthen their research and development, production, and marketing resources. Companies engaged in the medical products
industry face the risk of obsolescence of their products and technologies as more advanced or cost-effective products and
technologies are developed by competitors. As the industry matures, companies will compete based upon the performance and
cost-effectiveness of their products.
Products for Regenerative Medicine
The cell therapy industry is characterized by
rapidly evolving technology and intense competition. Kadimastem’s competitors include major multinational pharmaceutical companies,
specialty biotechnology companies, and chemical and medical products companies operating in the fields of regenerative medicine, cell
therapy, tissue engineering, and tissue regeneration. Many of these companies are well established and possess technical, research and
development, financial, and sales and marketing resources significantly greater than Kadimastem’s. In addition, certain smaller
biotech companies have formed strategic collaborations, partnerships, and other types of joint ventures with larger, well-established
industry competitors that afford the smaller companies’ potential research and development as well as commercialization advantages.
Academic institutions, governmental agencies, and other public and private research organizations are also conducting and financing research
activities, which may produce products directly competitive to those Kadimastem is developing.
Potential Competition for the AstroRx®
product
To the best of Kadimastem’s knowledge, there
is currently no cell/astrocytes-based therapy that has been differentiated from pluripotent stem cells for the treatment of ALS approved
for marketing by the FDA, the European Medicines Agency, or EMA, or any other jurisdiction. ALS is a multifactorial disease, characterized
by a progressive loss of motor neurons that eventually leads to paralysis and death. The current ALS-approved drugs modestly change the
clinical course of the disease. The contribution of astrocytes to the maintenance of motor neurons by diverse mechanisms makes them a
promising therapeutic candidate for the treatment of ALS. Therapeutic approaches targeting at modulating the function of endogenous
astrocytes or replacing lost functionality by transplantation of healthy astrocytes, may contribute to the development of therapies which
might slow down or even halt the progression of ALS diseases.
MSCs are adult multipotent-precursors that can
be derived from bone-marrow or placenta, with the potential to differentiate into osteocytes, chondrocytes, fibroblasts, and adipocytes.
MSC are not natural residents of the CNS but can be induced to secrete some of the neurotrophic factors secreted by astrocytes. There
is a product based on mesenchymal stem cells that was approved through an expedited process in South Korea by Corestem, Inc. However,
to the best of Kadimastem’s knowledge, Corestem did not conduct an experiment and was unable to obtain regulatory approval outside
of Korea. In addition, to the best of Kadimastem’s knowledge, there are currently several medical applications based on stem cells
that are in various development processes mainly by academic principal investigator, and which the company considers to be direct potential
competitors to the medicine that Kadimastem is developing in the field of activity. These include Q Therapeutics, Neuralstem and NurOwn®
by Brainstorm Cell Therapeutics, Inc. NurOwn® is the only product of direct competition that reached high stages in a Phase 3
clinical trial. To the best of Kadimastem’s knowledge, this product was denied a Biological Licensing Approval, or BLA, an FDA marketing
approval of a biological drug, for ALS treatment in 2022. As of April 2023, the FDA has agreed to have an additional advisory meeting
to reconsider the marketing of this product to ALS patients. Since all the MSC product candidates do not replace astrocytes, but rather
have a few astrocytes-like attributes, Kadimastem considers them competition for the short run, but strongly believe it has a competitive
edge on them.
There are currently on the market several products
(not based on the use of cells) or under development for the treatment of ALS, which are in various stages of development, as well as
three products approved by the FDA: (1) Rilutek, which has been marketed since the 1990s; (2) Edaravone, which was approved
in mid-2017 as an intra venous agent and again in late 2022 as an oral drug (Marketed by Mitsubishi Tanabe Pharmaceuticals as Radicava®),
which, to the best of Kadimastem’s knowledge, each has limited efficacy; and (3) RELYVRIO® (sodium phenylbutyrate
and taurursodiol, AMX0035) which is marketed by Amylyx, which has been approved by the FDA and the Ministry of Health in Canada, and is
in the stages of evaluation by EMA regarding the granting of a marketing authorization for its use. Kadimastem sees these products as
potential indirect competition with the drug that Kadimastem is developing.
IsletRx — Development of
a Drug for the Treatment of Diabetes
Development of Cell Therapies in the Field
of Regenerative Medicine for the Treatment of Insulin-Dependent Diabetes
Diabetes is a chronic disease that occurs when
the insulin-secreting beta cells in the pancreas do not produce enough insulin (such as after the immune-mediated destruction of beta
cells in Type 1 diabetes), or when the body cannot effectively use the insulin the beta cells produce (such as in Type 2 diabetes due
to insulin resistance in liver, fat and muscle tissues). Type 2 diabetes accounts for about 90% to 95% of all diagnosed cases of diabetes;
Type 1 diabetes accounts for about 5% to 10%.
Kadimastem’s Solution
IsletRx is a cellular therapy product based on
pancreatic islet-like clusters, or ILCs, derived from human embryonic stem cells for the treatment of Type 1 diabetes and possibly also
for insulin-dependent Type 2 diabetes. The ILCs are differentiated stepwise from the GMP-grade HAD C-100 hESC line. At the end of the
differentiation protocol, the hESC-derived pancreatic ILCs contain mainly insulin secreting cells that have the capability to sense and
respond to alterations in glucose levels, with secretion of adequate amounts of insulin in direct response to glucose levels. Thus, IsletRx
has the potential to functionally replace patients’ malfunctioning Islet cells by secreting physiological levels of insulin to maintain
long-term continuous balanced glucose. IsletRx, as allogeneic islet cells for diabetic patients derived from an unlimited source of pluripotent
stem cells, can overcome the shortage in adequate biological material for islet transplantation therapy.
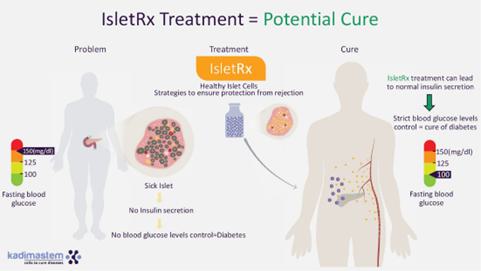
Conventional treatments (Insulin syringes and
pumps) are designed to monitor blood glucose levels in diabetics externally, and to inject insulin artificially according to the amount
needed (insulin is given directly into the blood by injections or by the pumps). This treatment has several disadvantages, including:
dependence on injections and external monitoring of blood glucose levels; the necessity to operate, maintain and fill the pumps, which
affects the patients’ quality of life; artificial injection of insulin, which does not prevent the deterioration of diabetes over
time; and excessive insulin injection, which may cause the glucose level to drop and lead to fainting, loss of consciousness and even
death in extreme situations. On the contrary, IsletRx has the potential to functionally replace patients’ malfunctioning Islet cells
by secreting the exact physiological levels of insulin to maintain long-term continuous balanced glucose levels with minimal risk, personal
comfort and increased compliance for treatment.
Strengths
Kadimastem’s vision is to utilize cell therapy
treatments for incurable diseases that impair patients’ life expectancy and quality of life, such as Type 1 diabetes. The strategy
is to replace the functionality of malfunctioning cells even within some distance from the original tissue/organ that is difficult or
impossible to replace. The activity of malfunctioning insulin secreting cells can be restored using “healthy” islet cells
that perform their physiological role upon their implantation.
Kadimastem believes they have the potential to
transform the lives of individuals living with Type 1 Diabetes and in the future Type 2 Diabetes which are insulin dependent.
Kadimastem’s key competitive strengths include:
| ● | Kadimastem developed large scale manufacturing capabilities
for IsletRx cells for clinical applications |
| ● | Kadimastem plans to utilize multiple delivery strategies,
developed in-house or in collaboration |
| ● | Kadimastem has a unique, IP protected technology for “enrichment”
of the therapeutically relevant pancreatic islet cells that it believes will result in increased safety and efficacy profile |
IsletRx — Development of a
Drug for the Treatment of Diabetes
The IsletRx cells for diabetes therapy Kadimastem
develops is based on differentiation of pluripotent stem cells received under license agreements. Kadimastem developed technologies for
the cultivation of pluripotent stem cells and their differentiation into islet-like cell clusters (containing mainly insulin-producing
beta cells and to lesser extent glucagon-producing alpha cells). These cells are intended for transplantation into Type 1 diabetic patients
whose beta cells are unable to produce insulin and release it in response to an increase in blood sugar, as well as adequate levels of
active glucagon that can respond to a sharp drop in glucose levels (hypoglycaemia). In this way, the transplant recipient can produce,
and release insulin or glucagon as needed, independently of external monitoring of blood sugar and regular insulin injection. The drug
Kadimastem is developing is intended for patients with Type 1 diabetes and in the future also for patients with Type 2 diabetes who are
dependent on injecting insulin from an external source to maintain adequate levels of sugar (glucose) in the blood.
Patients with uncontrolled diabetes suffer from
long-term complications such as vascular damage, decreased kidney function (leading to kidney failure), impaired vision and the development
of necrosis in the limbs. Therefore, by achieving strict management of blood sugar levels, the deterioration of the disease complications
can be avoided. In addition, the patient’s quality of life will be improved: at current state, the patient must perform regular
tests and carefully manage energy intake to prevent events of hypoglycaemia (very low, abnormal blood sugar level) resulting from excess
of injected external source insulin, or events of hyperglycaemia (very high, abnormal blood sugar level) resulting from insufficient insulin
injected from an external source.
For the development of IsletRx cells, Kadimastem
relies on these main technologies:
Expansion: Kadimastem
has the technological ability to multiply/expand the pluripotent stem cells received under license agreements and to preserve them in
deep freezing, while maintaining their vitality and pluripotent fitness. This technology enables the production and storage of industrial-scale
stock of pluripotent stem cells, which can then be thawed and used in production and differentiated into specific cell lineage as needed.
Differentiation: Kadimastem
has developed technology and know-how, through which the technology can mimic the process of embryonic development and direct the pluripotent
stem cells to differentiate into a specific lineage in the body, according to choice and relevant needs. The differentiation process is
carried out under laboratory conditions, in several stages, by adding chemical and/or biological agents in controlled timings to the pluripotent
stem cell culture. Induction of differentiation processes of pluripotent stem cells into islet-like cell clusters, under laboratory conditions
is conducted in a stirred suspension bioreactor under conditions that mimic the development of the pancreatic islets.
Enrichment: Kadimastem
has developed an enrichment technology which it believes improves safety and efficacy of IsletRx cells. The IsletRx cells are enriched
for insulin secreting cells, designated as IsletRxPlus, and formulated as the drug substance.
Delivery: Several
delivery strategies were developed to obviate the need for life-long immunosuppressive treatment while enabling the long-term function
of IsletRxPlus cells. These strategies include immune tolerance, microencapsulation in polymer, and scaffolding using electrospinning
of polycaprolactone. The cellular component in all products is the same, IsletRxPlus cells.
Products description
iTOL-102 Human pluripotent (embryonic)
stem cell-derived pancreatic islet-like cell clusters (IsletRxPlus); mixed with SA-FasL Microgels (iTOL-100), intended for
omental implantation. The iTOL100 enables implanting of IsletRxPlus cells without the need of chronic immunosuppression treatment.
Thus, IsletRxPlus cells are in direct contact with the omental vasculature, ensuring proper engraftment and function. Preclinical
efficacy studies using iTOL-102 demonstrated prolonged anti-diabetic effect, with physiological secretion of human insulin in a glucose
dependent manner. These results, along with the proof of concept, or POC, studies with an iTOL100 component, have led to submission of
a Pre-IND package.
Encap-IsletRxPlus Human pluripotent
(embryonic) stem cell-derived pancreatic islet-like cell clusters (IsletRxPlus); encapsulated in alginate microgels, intended
for intra-periotenal implantation. The alginate microencapsulation enables implanting of IsletRxPlus cells without the need
of chronic immunosuppression treatment. Still, IsletRxPlus cells are not in direct contact with the vasculature, and may suffer
from hypoxia and loss of function due to fibrosis. The retrievability of microencapsulated IsletRxPlus cells is a regulatory
concern, as there is very limited data about the safety of stem cells-derived islets. Preclinical efficacy studies using Encap-IsletRxPlus
demonstrated prolong anti-diabetic effect, with physiological secretion of human insulin in a glucose dependent manner.
In-Scaffold-IsletRxPlus Human
pluripotent (embryonic) stem cell-derived pancreatic islet-like cell clusters (IsletRxPlus); encapsulated in alginate microgels
embedded in electro spun polycaprolactone membrane, intended for subcutaneous implantation. The alginate microencapsulation enables implanting
of IsletRxPlus cells without the need of chronic immunosuppression treatment. The embedding of the microcapsules in a membrane
makes it a retrievable solution and obviates the regulatory concern. POC studies using In-Scaffold-IsletRxPlus demonstrated
that microencapsulated IsletRxPlus cells maintain their identity and capability to secrete insulin in immunodeficient mice.
Next planned studies aim to test the anti-diabetic effect of this delivery method. Kadimastem expects to submit a pre-IND package in the
second quarter of 2025.
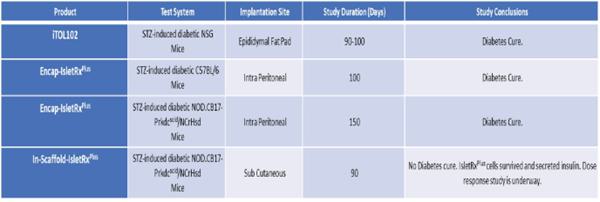
These delivery options are in various development
stages. The most advanced is iTOL-102, currently at the Pre-IND stage. Preclinical development studies were conducted to support regulatory
submissions for iTOL-102 (Pre-IND) and Encap-IsletRxPlus (INTERACT).
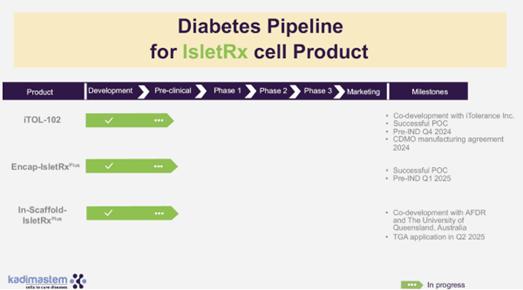
Drug administration strategies
The use of allogeneic islets in clinical settings
requires to ensure their long-term function in the patient without being destroyed by patient’s immune system Kadimastem utilizes
several technologies to protect the islets from the recipient’s immune system, which will eliminate the implant rejection risk,
without the need to administer immunosuppressive drugs to weaken the patient’s immune system and ensure long term function of IsletRxPlus
cells.
iTOL-102
iTOL-102 are human pluripotent (embryonic) stem-cell
derived islet-like clusters mixed with iTOL-100 (biotinylated PEG microgels with surface bound SA-FasL). iTOL-102 consists of IsletRxPlus
cells and iTOL-100 prepared for delivery into the omentum, a part of the stomach of diabetic patients. The therapeutic goal for iTOL-102
is to provide an easily administered, low risk, treatment via a minimally invasive procedure with long-term glucose responsiveness, that
can treat diabetes without the need for chronic immunosuppression or exogenous insulin supplementation. Data from initial proof-of-concept
studies with iTOL-102 in diabetic mouse models demonstrated potential efficacy and safety results of iTOL-102. In January 2024, iTolerance
and Kadimastem held a meeting with the FDA (INTERACT) to discuss safety, functionality and planning regarding the development of the joint
iTOL-102 project. In June 2024, The Diabetes Research Institute at the University of Miami Miller School of Medicine presented the results
of iTOL-102 POC study in the American Diabetes Association’s 84th Scientific Sessions that highlights the potential of human, stem
cell-derived islets combined with an immunomodulatory microgel to reverse Type 1 diabetes (T1D). The results demonstrated that the combination
of iTOL-100 engineered microgel developed by iTolerance and IsletRx stem cell-derived islets developed by Kadimastem, iTOL-102, can restore
normoglycemia in a model of diabetes. Accordingly, the companies are working on submitting a pre-IND package in the second quarter of
2025.

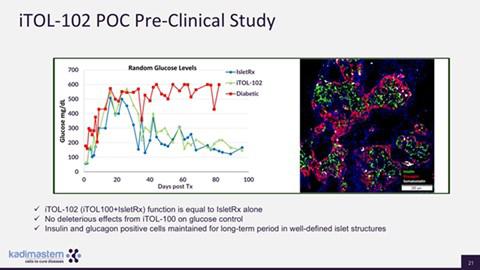
Encap-IsletRxPlus
Microencapsulation of cells within semi-permeable
hydrogels represents an immune isolation strategy for cell-based therapies without the need for systemic immunosuppression. Studies have
demonstrated that the microencapsulation process does not hinder the function of islets, and the insulin is efficiently secreted in response
to elevated blood glucose levels, using similar doses of islets as used in the Edmonton protocol. Preclinical studies have demonstrated
long-term Encap-IsletRxPlus functionality, in terms of continuous balanced glucose levels, in immunocompetent mice without immunosuppressive
treatment. The microencapsulation provides a physical barrier that protects the IsletRxPlus from the host immune system. Kadimastem’s
aim for Encap-IsletRxPlus is an easily administered, low risk, minimally invasive procedure in which encapsulated islets with long-term
glucose responsiveness can treat diabetes without the need for immunosuppression or exogenous insulin supplementation. A pre-IND submission
is planned for the second quarter of 2025, respectively Encap-IsletRxPlus.
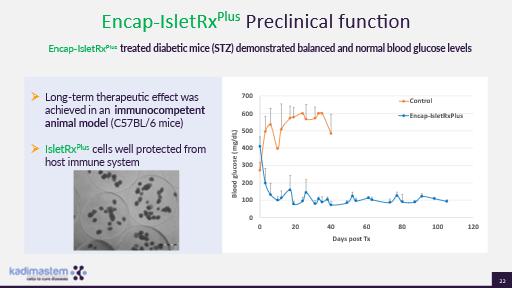
In-Scaffold-IsletRxPlus
Although alginate microcapsules provide immuno-isolation,
their surgical retrievability might be difficult. To ensure removal of IsletRx cells if needed, it is possible to implant them in a bioengineered
device to enable it. The device is a 3D printed scaffold containing encapsulated IsletRxPlus cells. The 3D printing process, called melt
electrospinning writing is a relatively novel solvent-less process that enables the design and fabrication of micrometer-thin fibers with
highly controllable architectures and patterns manufactured at the University of Queensland, Australia. Preclinical results demonstrated
that encapsulated islets placed in scaffolds and implanted subcutaneously or intraperitoneally,
produced human insulin and maintained viability
and identity for study duration (~3 months). Current Future studies aim to optimize this delivery strategy. In these studies, a dose
response study is planned, to analyze the minimal dose needed to achieve the desired effect, which is normal levels of glucose.
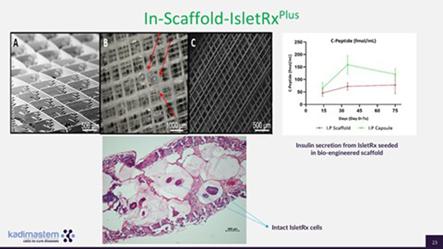
Key Commercial Agreements
License Agreement with Yeda Research and Development
Ltd.
In August 2009, Kadimastem entered into
a license agreement with Yeda Research and Development Ltd., or Yeda, a subsidiary of the Weizmann Institute, or the License
Agreement, under which Kadimastem received a license to use their invention, patents and knowledge for the purposes of developing,
manufacturing, marketing and selling stem cell-based drugs and treatments for diabetes and neuronal diseases, and in providing
various drug development services, including drug scanning. The agreement stipulates payments to be made to Yeda in accordance with
certain milestones as well as royalty payments beginning from the first commercial sale of a product, at a royalty rate of 10% to
14% of the “Net Sales” (as defined in the License Agreement) of product sold. The royalty payments expire upon the
earlier of 15 years from the date of the first sale of a licensed product under the License Agreement, and the expiration of
Yeda’s licensed patents under the License Agreement (the “Yeda IP”). The Yeda IP expires on separate dates; on
August 15, 2027, December 16, 2027, April 13, 2029, November 28, 2036, and July 23, 2038, each as detailed further below under
“— Intellectual Property” beginning on page 148.
Specifically, with respect to milestones reached,
Kadimastem will pay the following: (a) annual license fees in the amount of $20,000, which Kadimastem began paying from the fourth
year of the agreement; (b) a total of $650,000 after reaching sales of products (by Kadimastem and/or sublicensees, cumulatively)
in the amount of $100,000,000 (net); (c) a total of $2,500,000 after reaching such sales of goods in a cumulative amount of $500,000,000
(net); and (d) a total of $6,500,000 after reaching such sales of products in a cumulative amount of $5,000,000,000 (net). The agreement
terminates upon the expiration of Yeda’s patents licensed to Kadimastem under the agreement, and at Yeda’s option upon (a) Kadimastem
to meet certain Milestones (as defined below); and (b), a period of six (6) months of no sales of any product developed under the
agreement, or a “Product”. The Milestones include (a) within 2 (two) years from Agreement, Kadimastem must commence
pre-clinical activities directed toward the submission of an IND with respect to a Product; (b) within 3 (three) years from
the commencement of pre-clinical activities, to file an IND (or any equivalent application) with respect to a Product; (c) within
3 (three) years from IND filing, to commence a phase I clinical trial; (d) within 4 (four) years from the commencement
of a phase I clinical trial, to commence a phase III clinical trial; (e) within 4 (four) years from the commencement
of a phase III clinical trial, to receive regulatory approval for commencement of sales. As of the date of this annual report, $126,828
have been paid by Kadimastem to Yeda under this agreement.
Agreement with Ramot
In January 2015, Kadimastem entered into
a research and licensing agreement with Ramot for the commercialization arm of Tel Aviv University, and Professor Shimon Efrat, a leading
scientist in the joint research on behalf of Tel Aviv University. For the cooperation between the parties, Kadimastem must pay to Ramot
royalties at a rate of 0.3% – 0.8% for product sales, depending on progress according to the product’s regulatory
milestones and the degree of use of
Ramot’s technology developed during the
project, as stipulated in the agreement. In addition, royalties will be paid to Ramot at a rate of 2.75% for consideration received by
Kadimastem for sublicensing of product in the field of diabetes, depending on the milestone specified in the agreement; or royalties at
the rate of 7% if the sublicensing is for a product that combines the technology of Ramot. It was also agreed that Kadimastem would grant
to Ramot up to 200,000 options, free of charge, subject to reaching regulatory milestones, at the following rates: 30% of the options
would be granted after meeting with a regulatory body, prior to submitting the IND application; an additional 30% of the options will
be granted after the submission of the IND application; and an additional 40% of options will be granted after receiving the IND approval.
The granting of such options is subject to Professor Efrat continuing to advise Kadimastem throughout various stages of the product’s
regulatory approval. The agreement may be terminated at any time if either party commits a material breach or if either party goes into
bankruptcy, liquidation, makes a general assignment for the benefit of its creditors, or becomes a party to dissolution proceedings.
Feasibility Study Agreement with Australian
Foundation for Diabetes Research
Originally on September 2018, and as
further amended, Kadimastem entered into a feasibility study agreement with the Australian Foundation for Diabetes Research, or the
AFDR, and the Sydney Cell Therapy Foundation Pty Limited, or the SCTF, to evaluate the feasibility of implementing medical solutions
derived from Kadimastem’s human stem cell technologies into AFDR’s medical devices, licensed to SCTF. Pursuant to
this agreement, Kadimastem and AFDR jointly own, in equal parts of all intellectual property resulting from the collaboration and
share equally in the costs of patent filings and protections. In addition, Kadimastem received an exclusive, perpetual, worldwide
license to commercialize the joint results. As consideration for such license, Kadimastem will pay AFDR 22.5% of all
non-sales-related upfront payments it or its affiliates actually received from any sublicensee, and 25% of all milestone payments
actually received upon the occurrence or achievement of milestones by any sublicensee. The agreement terminates on December 31,
2026, or upon the execution of a commercialization agreement. The agreement may be terminated at any time if either party commits a
material breach or if either party goes into bankruptcy, liquidation, makes a general assignment for the benefit of its creditors,
or becomes a party to dissolution proceedings.
Cooperation and Project Funding Agreement with
iTolerance, Inc.
On May 23, 2023, Kadimastem, the Israel-United States
Binational Industrial Research and Development Foundation, or the Foundation, and iTolerance, Inc., or iTolerance, entered into a Pharma
Cooperation and Project Funding Agreement for the clinical development of iTol-102, or the Proposal, and collectively, the iTolerance
Co-Development Agreement. Under the iTolerance Co-Development Agreement, the Foundation agreed to fund the Proposal of up to $1,000,000,
or the Foundation Grant, with repayment made from any gross revenues received from any products developed under the Proposal, or a Product.
Following the earlier of (1) one year following an IND approval (or equivalent to any regulatory agency); (b) commencement of
the marketing of any Product; or (c) the continuation of the development of the Product, the Foundation Grant shall become immediately
payable to the Foundation, with the amounts to be repaid each year subject to certain maximum limits on the amount of gross sales to be
repaid each year. Under the iTolerance Co-Development Agreement, the governments of Israel and the United States were granted a non-exclusive,
irrevocable, royalty-free license to use any product for any purpose. The Foundation has the right to revoke the Foundation Grant upon
the occurrence, among other events, a fundamental breach of the agreement, insolvency, change in business, dispute between, or material
advance change in financial position of either Kadimastem or iTolerance.
Israel Innovation Authority Grant Framework
Under a general framework agreement with the IIA,
Kadimastem received approximately $13.9 million in grants, or the Grant, from the IIA under Incentive Track No. 1 — The
R&D Fund, or the Fund, under which it agreed to pay royalties ranging from 3% – 3.5% on sales derived from products
developed with grant funding received from the Fund, as long as total sales are under $70 million, or 5% of such sales if they exceed
$70 million, until the total Grant is repaid (subject to inflation and LIBOR interest). The IIA retains the right to terminate the
Grant and demand repayment of all benefits if it deems the project’s likelihood of success are too low, unless Kadimastem demonstrates
diligent efforts to advance the Grant’s objectives. Any sale or transfer of intellectual property outside of Israel requires approval
from the IIA. All products developed by Kadimastem developed as a result of the Grant program remain subject to royalties on revenues,
including related services and revenues of affiliates or licensees, and any amounts collected by the IIA under these provisions are reinvested
to promote further technological innovation in Israel.
Clinical Trial Agreement with Hadasit Medical
Research Services and Development Ltd.
On March 2018, Kadimastem entered into a
clinical trial agreement with Hadasit Medical Research Services and Development Ltd., a wholly-owned subsidiary of Hadassah Medical Organization,
or Hadasit, and Dr. Marc Gotkine, or the Investigator, to evaluate the safety, tolerability, and therapeutic effects of transplanting
allogeneic astrocytes derived from human embryonic stem cells in patients with ALS, or the Clinical Trial. As consideration for the Clinical
Trial, Kadimastem will pay Hadasit a start-up, nonrefundable fee of $3,000 and an advance of $60,000, which will be deducted from the
final monthly payment. Additional monthly payments will be based on the actual visits, tests, and procedures performed each quarter. Kadimastem
is also responsible for any Hadassah Ethical Committee IRB fees. The agreement shall terminate upon completion of the Clinical Trial.
Either party may terminate the agreement if any party files a petition for its winding up, liquidation, or the appointment of a receiver
over a substantial portion of its assets. Kadimastem may terminate the agreement for any reason by providing a sixty days’
notice, and Hadasit may terminate the agreement if regulatory approvals for the Clinical Trial are revoked. Upon termination, Kadimastem
will pay Hadasit for work performed and any non-cancelable commitments incurred up to the termination date, and Hadasit must return any
unexpended funds. Termination does not affect Kadimastem’s intellectual property rights. As of the date of this annual report, $2,113,661
have been paid by Kadimastem to Hadasit under this agreement.
Intellectual Property
Kadimastem seeks and have obtained some patent
protection as well as other effective intellectual property rights for Kadimastem’s products and technologies in the United States
and internationally. Kadimastem’s policy is to pursue, maintain and defend intellectual property rights developed internally and
to protect the technology, inventions and improvements that are commercially important to the development of its business.
A list of each of Kadimastem’s patent families,
including each of their status, their filing locations, and application information, is set forth below:
| METHODS OF GENERATING GLIAL AND NEURONAL CELLS |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry Date |
|
Filing
Date
Application
No. |
|
Publication
Date + No. Issue
Date + Patent No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Expiration
Date |
45463
2006-075 |
|
Israel NP |
|
August 28,
2006
60/840,426 |
|
February 26, 2009 |
|
August 15,
2007
197291 |
|
June 27, 2013
197291
September 28, 2013
197291 |
|
Methods of generating glial cells |
|
Granted |
|
Yeda Research and Development Co. Ltd. REVEL Michel; CHEBATH Judith; IZRAEL Michal; KAUFMAN Rosalia LICENSE |
|
August 15,
2027 |
45464
2006-075 |
|
USA NP |
|
August 28,
2006
60/840,426 |
|
February 26, 2009 |
|
August 15,
2007
12/310,495 |
|
January 7, 2010
2010-0003751-A1
August 19, 2014
8,809,052 |
|
An in vitro method of generating mature branched oligodendrocytes which produce myelin |
|
Granted |
|
Yeda Research and Development Co. Ltd. REVEL Michel; CHEBATH Judith; IZRAEL Michal; KAUFMAN Rosalia LICENSE |
|
August 15,
2027 |
METHODS OF GENERATING GLIAL AND NEURONAL CELLS AND USE OF SAME FOR THE TREATMENT
OF MEDICAL CONDITIONS OF THE CNS |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry Date |
|
Filing
Date
Application
No. |
|
Publication
Date + No. Issue
Date + Patent
No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
84030
2006-075 |
|
USA CON |
|
August 28,
2006
60/840,426 |
|
July 11,
2014 |
|
August 15,
2007
14/329,510 |
|
October 30, 2014
2014-0322179-A1
April 25, 2017
9,631,175 |
|
A method of enhancing myelination in the CNS of a subject |
|
Granted |
|
Yeda Research and Development Co. Ltd. REVEL Michel; CHEBATH Judith; IZRAEL Michal; KAUFMAN Rosalia LICENSE |
|
December 13,
2027 |
METHODS OF GENERATING GLIAL AND NEURONAL CELLS AND USE OF SAME FOR THE TREATMENT
OF MEDICAL CONDITIONS OF THE CNS |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry Date |
|
Filing
Date
Application
No. |
|
Publication
Date + No. Issue
Date + Patent
No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
84031
2006-075 |
|
USA DIV |
|
August 28,
2006
60/840,426 |
|
March 23,
2017 |
|
August 15,
2007
15/468,020 |
|
December 21, 2017
2017-0362570-A1
April 16, 2019
10,258,651 |
|
A method of treating one or more symptoms of ALS in a subject in need thereof |
|
Granted |
|
Yeda Research and Development Co. Ltd. REVEL Michel; CHEBATH Judith; IZRAEL Michal; KAUFMAN Rosalia LICENSE |
|
April 13,
2029 |
84032
2006-075 |
|
USA CON |
|
August 28, 2006 60/840,426 |
|
February 21,
2019 |
|
August 15,
2007
16/281,490 |
|
June 13, 2019
2019-0175658-A1
August 9, 2022
11,406,670 |
|
An in vitro method of producing astrocytes |
|
Granted |
|
Yeda Research and Development Co. Ltd. REVEL Michel; CHEBATH Judith; IZRAEL Michal; KAUFMAN Rosalia LICENSE |
|
August 15,
2027 |
45462
2006-075 |
|
Europe NP |
|
August 28,
2006
60/840,426 |
|
March 27,
2009 |
|
August 15,
2007
07790074.4 |
|
June 3, 2009
2064319
February 22, 2017
2064319 |
|
In vitro methods of generating glial cells and neurons |
|
Granted |
|
Yeda Research and Development Co. Ltd. REVEL Michel; CHEBATH Judith; IZRAEL Michal; KAUFMAN Rosalia LICENSE |
|
August 15,
2027 |
47734
2006-075 |
|
Hong Kong NP |
|
August 28,
2006
60/840,426 |
|
December 2,
2009 |
|
August 15,
2007
09111292.3 |
|
January 29, 2010
1131640A
March 29, 2018
HK1131640 |
|
In vitro methods of generating glial cells and neurons |
|
Granted |
|
Yeda Research and Development Co. Ltd. REVEL Michel; CHEBATH Judith; IZRAEL Michal; KAUFMAN Rosalia LICENSE |
|
August 15,
2027 |
| INSULIN PRODUCING CELLS DERIVED FROM PLURIPOTENT STEM CELLS |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry Date |
|
Filing
Date
Application
No. |
|
Publication
Date + No.
Issue
Date + Patent
No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Parent
Expiration
Date |
| 84034 |
|
Canada
NP |
|
December 15,
2010 61/423,171 |
|
April 30,
2013 |
|
December 15,
2011 2,816,495 |
|
October 13, 2020
2,816,495 |
|
Methods of generating islet cells or islet progenitor cells from pluripotent stem cells and compositions comprising cells generated thereby and uses thereof |
|
Granted |
|
Kadimastem Ltd. REVEL Michel; CHEBATH Judith; SLUTSKY Guy; LEVY Alon; IZRAEL Michal; HASSON Arye; MOLAKANDOV Kfir; KAUFMAN Rosalia OWNERSHIP |
|
December 15,
2031 |
| 84035 |
|
Australia NP |
|
December 15,
2010 61/423,171 |
|
May 1, 2013 |
|
December 15,
2011 2011342732 |
|
August 11, 2016
2011342732 |
|
Methods of generating islet cells or islet progenitor cells from pluripotent stem cells and compositions comprising cells generated thereby and uses thereof |
|
Granted |
|
Kadimastem Ltd. REVEL Michel; CHEBATH Judith; SLUTSKY Guy; LEVY Alon; IZRAEL Michal; HASSON Arye; MOLAKANDOV Kfir; KAUFMAN Rosalia OWNERSHIP |
|
December 15,
2031 |
| INSULIN PRODUCING CELLS DERIVED FROM PLURIPOTENT STEM CELLS |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry Date |
|
Filing
Date
Application
No. |
|
Publication
Date + No.
Issue
Date + Patent
No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Parent
Expiration
Date |
| 84037 |
|
Europe
NP |
|
December 15,
2010 61/423,171 |
|
May 2, 2013 |
|
December 15,
2011 11848280.1 |
|
October 23, 2013
2652125
April 26, 2017
2652125 |
|
Methods of generating islet cells from pluripotent stem cells |
|
Granted |
|
Kadimastem Ltd. REVEL Michel; CHEBATH Judith; SLUTSKY Guy; LEVY Alon; IZRAEL Michal; HASSON Arye; MOLAKANDOV Kfir; KAUFMAN Rosalia OWNERSHIP |
|
December 15,
2031 |
| 84045 |
|
USA NP |
|
December 15,
2010 61/423,171 |
|
May 27, 2013 |
|
December 15,
2011 13/989,805 |
|
October 17, 2013
2013-0273013-A1
August 2, 2016
9,404,087 |
|
Methods of generating islet cells or islet progenitor cells from pluripotent stem cells and compositions comprising cells generated thereby and uses thereof |
|
Granted |
|
Kadimastem Ltd. REVEL Michel; CHEBATH Judith; SLUTSKY Guy; LEVY Alon; IZRAEL Michal; HASSON Arye; MOLAKANDOV Kfir; KAUFMAN Rosalia OWNERSHIP |
|
April 5,
2032 |
| 84046 |
|
Israel NP |
|
December 15,
2010 61/423,171 |
|
May 28, 2013 |
|
December 15,
2011
226617 |
|
January 31, 2019
226617
May 1, 2019
226617 |
|
Methods of generating islet cells or islet progenitor cells from pluripotent stem cells and compositions comprising cells generated thereby and uses thereof |
|
Granted |
|
Kadimastem Ltd. REVEL Michel; CHEBATH Judith; SLUTSKY Guy; LEVY Alon; IZRAEL Michal; HASSON Arye; MOLAKANDOV Kfir; KAUFMAN Rosalia OWNERSHIP |
|
December 15,
2031 |
| METHOD OF IDENTIFYING AGENTS THAT AFFECT MATURATION, SURVIVAL AND MYELINATION |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry Date |
|
Filing
Date
Application No. |
|
Publication
Date + No. Issue
Date + Patent No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
| 84069 |
|
Israel NP |
|
August 7,
2011
61/515,926 |
|
January 21,
2014 |
|
August 5,
2012
230576 |
|
March 30, 2017 230576
July 1, 2017
230576 |
|
An in vitro method of quantifying an effect of an agent on myelination of neuronal cells |
|
Granted |
|
Kadimastem Ltd. REVEL Michel; CHEBATH Judith; HASSON Arye; IZRAEL Michal; KAUFMAN Rosalia OWNERSHIP |
|
August 5,
2032 |
DIRECTED DIFFERENTIATION OF ASTROCYTES FROM HUMAN PLURIPOTENT STEM CELLS FOR USE IN DRUG
SCREENING AND THE TREATMENT OF AMYOTROPHIC LATERAL SCLEROSIS (ALS) |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry Date |
|
Filing
Date
Application No. |
|
Publication
Date + No. Issue
Date + Patent No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
| 84077 |
|
Israel NP |
|
October 1,
2013
61/885,018 |
|
March 29,
2016 |
|
September 23,
2014
244808 |
|
March 25, 2021 244808
June 26, 2021
244808 |
|
A method of screening an agent for preventing or treating ALS. A composition comprising non-genetically modified cell population of human progenitor astrocytes or astrocytes, for use in the treatment or prevention the progression of ALS |
|
Granted |
|
Kadimastem Ltd. IZRAEL Michal; REVEL Michel; HASSON Arye; MOLAKANDOV Kfir; CHEBATH Judith OWNERSHIP |
|
September 23,
2034 |
DIRECTED DIFFERENTIATION OF ASTROCYTES FROM HUMAN PLURIPOTENT STEM CELLS FOR USE IN DRUG
SCREENING AND THE TREATMENT OF AMYOTROPHIC LATERAL SCLEROSIS (ALS) |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry
Date |
|
Filing
Date
Application No. |
|
Publication
Date + No. Issue
Date + Patent No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
| 84749 |
|
USA CON |
|
October 1,
2013
61/885,018 |
|
September 9,
2019 |
|
September 23,
2014
16/564,893 |
|
February 25, 2021
2021-0054334-A1 |
|
A method of treating ALS comprising intrathetically
administering to a subject in need thereof non-genetically modified cell population of human progenitor astrocytes or astrocytes |
|
Pending |
|
Kadimastem Ltd. IZRAEL Michal; REVEL Michel; HASSON
Arye; MOLAKANDOV Kfir; CHEBATH Judith OWNERSHIP |
|
September 23,
2034 |
| 84080 |
|
Australia
NP |
|
October 1,
2013
61/885,018 |
|
April 1,
2016 |
|
September 23,
2014
2014330807 |
|
October 15,
2020
2014330807 |
|
A method of screening
an agent for preventing or treating ALS. A method for treating or preventing the progression of ALS in a subject in need thereof |
|
Granted |
|
Kadimastem Ltd.
IZRAEL Michal; REVEL Michel; HASSON Arye; MOLAKANDOV Kfir; CHEBATH Judith OWNERSHIP |
|
September 23,
2034 |
| 84752 |
|
Australia
DIV |
|
October 1,
2013
61/885,018 |
|
October 1,
2020 |
|
September 23,
2014
2020244551 |
|
November 24,
2022
2020244551 |
|
A method of treating or preventing
progression of ALS. in a human subject in need thereof |
|
Granted |
|
Kadimastem Ltd. IZRAEL Michal;
REVEL Michel; HASSON Arye; MOLAKANDOV Kfir; CHEBATH Judith OWNERSHIP |
|
September 23,
2034 |
| 84081 |
|
Canada
NP |
|
October 1,
2013 61/885,018 |
|
April 1,
2016 |
|
September 23,
2014
2,926,180 |
|
|
|
Use of a non-genetically modified
cell population of mature human astrocytes for the manufacture of a medicament for treatment of ALS |
|
Pending |
|
Kadimastem Ltd. IZRAEL Michal;
REVEL Michel; HASSON Arye; MOLAKANDOV Kfir; CHEBATH Judith OWNERSHIP |
|
September 23,
2034 |
DIRECTED DIFFERENTIATION OF ASTROCYTES FROM HUMAN PLURIPOTENT STEM CELLS FOR USE IN DRUG
SCREENING AND THE TREATMENT OF AMYOTROPHIC LATERAL SCLEROSIS (ALS) |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry
Date |
|
Filing
Date
Application No. |
|
Publication
Date + No. Issue
Date + Patent No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
| 84083 |
|
Europe
NP |
|
October 1,
2013
61/885,018 |
|
April 1,
2016 |
|
September 23,
2014
14851149.6 |
|
August 10,
2016
3052191
March 18, 2020
3052191 |
|
A method of screening an agent
for preventing or treating ALS. |
|
Granted |
|
Kadimastem Ltd. IZRAEL Michal;
REVEL Michel; HASSON Arye; MOLAKANDOV Kfir; CHEBATH Judith OWNERSHIP |
|
September 23,
2034 |
| 84086 |
|
Japan
NP |
|
October 1,
2013
61/885,018 |
|
April 1,
2016 |
|
September 23,
2014 2016-520086 |
|
February 27,
2019
6,474,795
February 8, 2019
6,474,795 |
|
A method for screening a drug
for preventing or treating ALS. |
|
Granted |
|
Kadimastem Ltd. IZRAEL Michal;
REVEL Michel; HASSON Arye; MOLAKANDOV Kfir; CHEBATH Judith OWNERSHIP |
|
September
23,
2034 |
| 84107 |
|
Japan
DIV |
|
October 1,
2013
61/885,018 |
|
August 15,
2018 |
|
September 23,
2014 2018-152497 |
|
November 25,
2021
6,983,128 |
|
Use of a non-genetically modified
cell population of mature human astrocytes for the manufacture of a medicament for treatment of ALS |
|
Granted |
|
Kadimastem Ltd. IZRAEL Michal;
REVEL Michel; HASSON Arye; MOLAKANDOV Kfir; CHEBATH Judith OWNERSHIP |
|
September
23,
2034 |
DIRECTED DIFFERENTIATION OF ASTROCYTES FROM HUMAN PLURIPOTENT STEM CELLS FOR USE IN DRUG
SCREENING AND THE TREATMENT OF AMYOTROPHIC LATERAL SCLEROSIS (ALS) |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry
Date |
|
Filing
Date
Application No. |
|
Publication
Date + No. Issue
Date + Patent No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
101959
PCT-2ALS-CH |
|
China DIV |
|
October 1,
2013
61/885,018 |
|
November 19,
2024 |
|
September 23,
2014
202411652013.7 |
|
|
|
Use of a non-genetically modified
cell population of mature human astrocytes for the manufacture of a medicament for treatment of ALS |
|
Pending |
|
Kadimastem Ltd. IZRAEL Michal; REVEL
Michel; HASSON Arye; MOLAKANDOV Kfir; CHEBATH Judith OWNERSHIP |
|
September 23,
2034 |
| METHODS
OF INDUCING MYELINATION AND MATURATION OF OLIGODENDROCYTES |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry
Date |
|
Filing
Date
Application
No. |
|
Publication
Date + No.
Issue
Date + Patent
No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
| 84095 |
|
Europe NP |
|
June 2, 2014
62/006,285 |
|
September 28,
2016 |
|
June 2, 2015
15802806.8 |
|
April 5, 2017
3149155 September 23, 2020
3149155 |
|
An ex vivo method of induring maturation of oligodendrocytes,
compositions obtained thereby and use thereof. |
|
Granted |
|
Kadimastem Ltd. IZRAEL Michal; REVEL Michel; HASSON
Arye; CHEBATH Judith OWNERSHIP |
|
June 2,
2035 |
| METHODS
FOR DIFFERENTIATING AND PURIFYING PANCREATIC ENDOCRINE CELLS |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry
Date |
|
Filing
Date
Application
No. |
|
Publication
Date + No.
Issue
Date + Patent
No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
| 84105 |
|
Brazil NP |
|
November 30,
2015
62/260,669 |
|
May 29,
2018 |
|
November 28,
2016
BR 11 2018 01094 6 |
|
|
|
A method for enriching pancreatic endocrine cells Compositions
comprising cells obtained by the method. |
|
Pending |
|
Kadimastem Ltd.; Yeda Research and Development Co.
Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael; HASSON Arye CO OWNERSHIP |
|
November 28,
2036 |
| 84101 |
|
Israel
NP |
|
November 30,
2015
62/260,669 |
|
May 30,
2018 |
|
November 28,
2016
259702 |
|
June 1,
2022
259702
September 2, 2022
259702 |
|
A method for enriching pancreatic
endocrine cells with specific markers Compositions comprising cells obtained by the method and use thereof. |
|
Granted |
|
Kadimastem Ltd.; Yeda Research
and Development Co. Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael; HASSON
Arye CO OWNERSHIP |
|
November
28,
2036 |
| 92532 |
|
Israel
DIV |
|
November 30,
2015
62/260,669 |
|
May 24,
May-2022 |
|
November 28,
2016
293289 |
|
December 1,
2023
293289
March 4,2024
293289 |
|
A method for enriching
pancreatic endocrine cells with specific markers Compositions comprising cells obtained by the method and use thereof. |
|
Granted |
|
Kadimastem Ltd.;
Yeda Research and Development Co. Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael;
HASSON Arye CO OWNERSHIP |
|
November
28,
2036 |
| METHODS
FOR DIFFERENTIATING AND PURIFYING PANCREATIC ENDOCRINE CELLS |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry
Date |
|
Filing
Date
Application
No. |
|
Publication
Date + No.
Issue
Date + Patent
No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
| 84102 |
|
Japan NP |
|
November 30,
2015
62/260,669 |
|
May 30,
2018 |
|
November 28,
2016
2018-546931 |
|
July 6, 2021
6,909,230 |
|
A method for enriching pancreatic endocrine cells with
specific markers Compositions comprising cells obtained by the method and use thereof. |
|
Granted |
|
Kadimastem Ltd.; Yeda Research and Development Co.
Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael; HASSON Arye CO OWNERSHIP |
|
November 28,
2036 |
| 84103 |
|
USA NP |
|
November 30,
2015
62/260,669 |
|
May 30,
2018 |
|
November 28,
2016
15/780,153 |
|
December 27,
2018-0369290-A1
June 20, 2023
11,679,133 |
|
A method for enriching pancreatic
endocrine cells with specific markers |
|
Granted |
|
Kadimastem Ltd.; Yeda Research
and Development Co. Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael; HASSON
Arye CO OWNERSHIP |
|
July 23,
2038 |
| 96080 |
|
USA
CON |
|
November 30,
2015
62/260,669 |
|
April 21,
2023 |
|
November 28,
2016
18/137,455 |
|
August 10,
2023
2023-0248779-A1 |
|
A method for enriching
pancreatic endocrine cells with specific markers |
|
Pending |
|
Kadimastem Ltd.;
Yeda Research and Development Co. Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael;
HASSON Arye CO OWNERSHIP |
|
November
28,
2036 |
| METHODS
FOR DIFFERENTIATING AND PURIFYING PANCREATIC ENDOCRINE CELLS |
Our Ref
Client Ref |
|
Country |
|
Earliest
Priority |
|
Entry
Date |
|
Filing
Date
Application
No. |
|
Publication
Date + No.
Issue
Date + Patent
No. |
|
Subject
Matter |
|
Status |
|
Assignee
Inventor |
|
Patent
Expiration
Date |
| 84104 |
|
Europe
NP |
|
November 30,
2015
62/260,669 |
|
June 11,
2018 |
|
November 28,
2016
16870121.7 |
|
October 10,
2018
3384286
April 19, 2023
3384286 |
|
A method for enriching
pancreatic endocrine cells with specific markers |
|
Granted |
|
Kadimastem Ltd.;
Yeda Research and Development Co. Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael;
HASSON Arye CO OWNERSHIP |
|
December 28,
2036 |
| 95901 |
|
Europe
DIV |
|
November 30,
2015
62/260,669 |
|
March 15,
2023 |
|
November 28,
2016
23162158.2 |
|
August 23,
2023
4231015
October 30, 2024
4231015 |
|
A method for enriching pancreatic
endocrine cells with specific markers |
|
Granted |
|
Kadimastem Ltd.; Yeda Research
and Development Co. Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael; HASSON
Arye CO OWNERSHIP |
|
November 28,
2036 |
| 84392 |
|
India NP |
|
November 30,
2015
62/260,669 |
|
July 2,
2018 |
|
November 28,
2016
201817024538 |
|
December 7,
2023
478779 |
|
A method for enriching pancreatic
endocrine cells with specific markers |
|
Granted |
|
Kadimastem Ltd.; Yeda Research
and Development Co. Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael; HASSON
Arye CO OWNERSHIP |
|
November
28,
2036 |
| 99188 |
|
Hong Kong
DIV |
|
November 30,
2015
62/260,669 |
|
February 19,
2024 |
|
November 28,
2016
42024087278.8 |
|
March 28,
2024
HK40098139 |
|
A method for enriching
pancreatic endocrine cells with specific markers |
|
Allowed |
|
Kadimastem Ltd.;
Yeda Research and Development Co. Ltd. MOLAKANDOV Kfir; LAVON Neta; BECK Avital; REVEL Michel; ELHANANI Ofer; SOEN Yoav; WALKER Michael;
HASSON Arye CO OWNERSHIP |
|
November
28,
2036 |
Trademark: NUCELX
Kadimastem’s
Ref Client Ref |
|
Country |
|
Class |
|
Earliest
Priority |
|
Filing
Date
Application No. |
|
Registration
Date
Registration No. |
|
Next
Action |
|
Status |
|
Applicant |
| 102648 |
|
Israel |
|
|
|
|
|
|
|
|
|
|
|
Pending |
|
Kadimastem Ltd. |
Trademark: AstroRx
Kadimastem’s
Ref Client Ref |
|
Country |
|
Class |
|
Earliest
Priority |
|
Filing
Date
Application No. |
|
Registration
Date
Registration No. |
|
Next
Action |
|
Status |
|
Applicant |
| 99994 |
|
Israel |
|
|
|
|
|
July 26, 2016
286776 |
|
January 1, 2018
286776 |
|
Renewal July 26,
2026 |
|
Registered |
|
Kadimastem Ltd. |
Employees
As of December 31,
2024, Kadimastem had 10 employees, six of whom are employed on a full-time basis and four employed on a part-time basis. Five of Kadimastem’s
employees are in its research and development department and five employees are in its general and administrative department.
Israeli labor laws
principally govern the length of the workday, minimum wages for employees, procedures for hiring and dismissing employees, determination
of severance pay, annual leave, sick days, advance notice of termination of employment, equal opportunity and anti-discrimination
laws and other conditions of employment. Subject to certain exceptions, Israeli law generally requires severance pay upon the retirement,
death or dismissal of an employee, and requires Kadimastem and its employees to make payments to the National Insurance Institute, which
is similar to the U.S. Social Security Administration. Kadimastem employees have defined benefit pension plans that comply with
applicable Israeli legal requirements, which also include the mandatory pension payments required by applicable law and allocations for
severance pay.
While none of Kadimastem’s
employees are party to any collective bargaining agreements, certain provisions of the collective bargaining agreements between the Histadrut
(General Federation of Labor in Israel) and the Coordination Bureau of Economic Organizations (including the Industrialists’ Associations)
are applicable to Kadimastem employees by extension orders issued by the Israel Ministry of Economy (previously the Israeli Ministry
of Trade, Industry and Labor). These provisions primarily concern the length of the workweek, pension fund benefits for all employees
and for employees in the industry section, insurance for work-related accidents, travel expenses reimbursement, holiday leave, convalescent
payments and entitlement for vacation days. Kadimastem generally provides its employees with benefits and working conditions beyond
the required minimums. Kadimastem has never experienced any employment-related work stoppages and believes its relationship with its
employees is good.
All of Kadimastem’s
employment and consulting agreements include employees’ and consultants’ undertakings with respect to non-competition and
assignment to Kadimastem of intellectual property rights developed in the course of employment and confidentiality. The enforceability
of such provisions is limited by Israeli law.
Government Regulation
and Product Approval
Governmental authorities
in the United States and in other countries extensively regulate, among other things, the research, development, testing, manufacture,
labeling, packaging, promotion, storage, advertising, distribution, marketing and export and import of products such as those Kadimastem
is developing. Any product candidate must be approved by the FDA through the NDA process before they may be legally marketed in the United States
and by the Committee on Human Medicinal Products, or CHMP, via the EMA and European Commission through the MAA process before they may
be legally marketed in Europe, or MHRA through its authorization procedures before they may be legally marketed in the UK. Any product
candidate will be subject to similar requirements in other countries prior to marketing in those countries. The process of obtaining
regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations require
the expenditure of substantial time and financial resources.
United States
Government Regulation
NDA Approval
Processes
In the United States,
the FDA regulates drugs under the FDCA and implementing regulations and guidance documents. Failure to comply with the applicable U.S. requirements
at any time during the product development process or approval process, or after approval, may subject an applicant to administrative
or judicial sanctions, any of which could have a material adverse effect on Kadimastem. These sanctions could include refusal to approve
pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product seizures, total
or partial suspension of production or distribution, injunctions, fines, disgorgement, and civil or criminal penalties.
The process required
by the FDA before a drug may be marketed in the United States generally involves the following:
| ● | completion of pre-clinical laboratory
tests, animal studies and formulation studies conducted according to GLPs, or other applicable regulations; |
| ● | submission to the FDA of an
IND application, which must become effective before human clinical trials may begin; |
| ● | performance of adequate and
well-controlled human clinical trials according to GCPs, to establish the safety and efficacy of the proposed drug for its intended use; |
| ● | submission to the FDA of an
NDA; |
| ● | satisfactory completion of an
FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with cGMPs to assure
that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
| ● | satisfactory completion of FDA
inspections of clinical sites and GLP toxicology studies; and |
| ● | FDA review and approval of the
NDA. |
The
testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for
a product candidate will be granted on a timely basis, if at all.
Once a product
candidate is identified for development, it enters the pre-clinical testing stage. Pre-clinical tests include laboratory evaluations
of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the pre-clinical
tests, together with manufacturing information and analytical data, to the FDA as part of the IND. Some pre-clinical testing may
continue after the IND is submitted. In addition to including the results of the pre-clinical studies, the IND will also include a clinical
trial protocol detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and,
depending on the phase of the study, the effectiveness criteria to be evaluated. The IND automatically becomes effective 30 days
after receipt by the FDA, unless the FDA, within the 30-day time period, places the IND on clinical hold. In such a case, the IND sponsor
and the FDA must resolve any outstanding concerns before clinical trials can begin. A clinical hold may occur at any time during the
life of an IND, due to safety concerns or non-compliance, and may affect one or more specific studies or all studies conducted under
the IND.
All clinical trials
must be conducted under the supervision of one or more qualified investigators in accordance with the FDA’s GCP regulations. These
regulations include the requirement that all research subjects provide informed consent. Further, an IRB must review and approve the
plan for any clinical trial, including the informed consent document, before it commences at any institution. An IRB considers, among
other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated
benefits. The IRB also approves the investigator brochure and other information about the trial distributed by the sponsor and the consent
form that must be provided to each trial subject or his or her legal representative and must monitor the study until completed. All clinical
trials must be conducted under protocols detailing the objectives of the trial, dosing procedures, research subject inclusion and exclusion
criteria and the safety and effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND, and
progress reports detailing the status of the clinical trials must be submitted to the FDA annually. Sponsors must also report within
set timeframes to FDA serious and unexpected adverse reactions, any clinically important increase in the rate of a serious suspected
adverse reaction over that listed in the protocol or investigation brochure, or any findings from other studies or animal or in-vitro
testing that suggest a significant risk in humans exposed to the drug. Sponsors must also report to FDA certain amendments to the protocol
and other essential information concerning the IND that does not fall within the scope of other required reports.
Human clinical
trials are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase 1. The
drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution
and elimination. In the case of some products for severe or life-threatening diseases, such as cancer, especially when the product may
be inherently too toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| ● | Phase 2. Clinical
trials are performed on a limited patient population intended to identify possible adverse effects and risks, to preliminarily evaluate
the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| ● | Phase 3. Clinical
trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed
clinical study sites. Phase 3 clinical trials are conducted to provide sufficient data for the statistically valid evidence of safety
and efficacy. |
| ● | Phase 4. The
FDA may require that the sponsor conduct additional clinical trials following new drug approval. The purpose of these trials, known as
Phase 4 studies, is to monitor long-term risks and benefits, study different dosage levels or evaluate safety and effectiveness.
In recent years, the FDA has increased its reliance on these trials. Phase 4 studies usually involve thousands of participants.
Phase 4 studies also may be initiated by the company sponsoring the new drug to gain broader market value for an approved drug. |
Human clinical
trials are inherently uncertain and Phase 1, Phase 2, Phase 3 and Phase 4 testing may not be successfully completed.
The FDA or the sponsor may suspend a clinical trial at any time for a variety of reasons, including a finding that the research subjects
or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial
at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been
associated with unexpected serious harm to patients.
During the development
of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points are typically prior to the submission
of an IND, at the end of Phase 2 and before an NDA is submitted. Meetings at other times may also be requested. These meetings can
provide an opportunity for the sponsor to share information about the data gathered to date and for the FDA to provide advice on the
next phase of development. Sponsors typically use the meeting at the end of Phase 2 to discuss their Phase 2 clinical results
and present their plans for the pivotal Phase 3 clinical trial that they believe will support the approval of the NDA.
Concurrent with
clinical trials, sponsors usually complete any remaining animal safety studies and also develop additional information about the chemistry
and physical characteristics of the drug and finalize a process for manufacturing commercial quantities of the product in accordance
with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug and the manufacturer
must develop methods for testing the quality, purity and potency of the drug. Additionally, appropriate packaging must be selected and
tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over
its proposed shelf-life.
The results of
product development, pre-clinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests
and other control mechanisms, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting
approval to market the product for one or more specified indications. The submission of an NDA is subject to the payment of an application
fee, but a waiver of such fees may be obtained under specified circumstances. We will seek a waiver of these fees as a small business
submitting its first human drug application to the FDA. If the waiver is granted it would not extend to establishment or product
fees. The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them
for filing. It may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with
the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
Once the submission
is accepted for filing, the FDA begins an in-depth review. The FDA may refuse to approve an NDA if the applicable statutory and regulatory
criteria are not satisfied or may require additional clinical or other data. Even if such data are submitted, the FDA may ultimately
decide that the NDA does not satisfy the criteria for approval. The FDA reviews an NDA to determine, among other things, whether a product
is safe and effective for its intended use and whether its manufacturing is cGMP-compliant. The FDA may refer the NDA to an advisory
committee for review and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound
by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will
typically inspect the facility or facilities where the product is manufactured and tested. The FDA will also inspect selected clinical
sites that participated in the clinical studies and may inspect the testing facilities that performed the GLP toxicology studies cited
in the NDA.
Expedited Review
and Approval
The FDA has various
specific programs, including Fast Track, Breakthrough Therapy, Priority Review, and Accelerated Approval, which, in different ways, are
each intended to expedite the process for reviewing and approving drugs. Even if a drug qualifies for one or more of these programs,
the FDA may later decide that the drug no longer meets the conditions for qualification or that the time period for FDA review or approval
will be shortened. Generally, drugs that are eligible for these programs are those for serious or life-threatening conditions, those
with the potential to address unmet medical needs and those that offer meaningful benefits over existing treatments. For example, Fast
Track is a process designed to facilitate the development and expedite the review of drugs to treat serious or life-threatening diseases
or conditions and fill unmet medical needs, and Breakthrough Therapy designation is designed to expedite the development and review of
drugs that are intended to treat a serious condition where preliminary clinical evidence indicates that the drug may demonstrate substantial
improvement over available therapy on a clinically significant endpoint(s). Priority review is designed to give drugs that offer major
advances in treatment or provide a treatment where no adequate therapy exists an initial review within six months as compared to
a standard review time of ten months. Although Fast Track, Breakthrough Therapy designation and priority review do not affect the
standards for approval, the FDA will attempt to facilitate early and frequent meetings with a sponsor of a Fast Track or Breakthrough
Therapy designated drug and expedite review of the application for a drug designated for priority review. The FDA will also provide Breakthrough
Therapy designated drugs intensive guidance on an efficient drug development program and provide these drug developers with an organizational
commitment from the FDA involving senior managers. Since sponsors can design clinical trials in a number of ways, in providing its guidance
for drugs designated as breakthrough therapies, the FDA will seek to ensure that the sponsor of the product designated as a breakthrough
therapy receives timely advice and interactive communications in order to help the sponsor design and conduct a development program as
efficiently as possible. During these interactions, the FDA may suggest, or a sponsor can propose, alternative clinical trial designs
(e.g., adaptive designs, an enrichment strategy, use of historical controls) that may result in smaller trials or more efficient trials
that require less time to complete. Such trial designs could also help minimize the number of patients exposed to a potentially less
efficacious treatment (i.e., the control group treated with available therapy).
Accelerated Approval,
which is described in 21 C.F.R. § 314.500 et seq., provides for approval of a new drug that is intended to treat a serious
or life-threatening disease or condition and that fills an unmet medical need based on a surrogate endpoint. A surrogate endpoint is
a laboratory measurement or physical sign used as an indirect or substitute measurement representing a clinically meaningful outcome.
To be used in accelerated approval, a surrogate endpoint must be “reasonably likely, based on epidemiologic, therapeutic, pathophysiologic,
or other evidence to predict benefit on irreversible morbidity or mortality.” The term “reasonably likely” implies
that some uncertainty remains about the relationship of the surrogate to the clinical benefit to the patient. Therefore, accelerated
approval is typically contingent on a sponsor’s agreement to conduct additional post-approval studies to verify and describe the
drug’s clinical benefit. Accelerated Approval does not change the standards for approval, but by allowing a demonstration of efficacy
based on a surrogate endpoint may expedite the approval process.
Patent Term
Restoration and Marketing Exclusivity
Depending upon
the timing, duration and specifics of FDA approval of the use of any product candidate, U.S. patents may be eligible for limited
patent term extension under the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as
compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration
cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term
restoration period is generally one-half the time between the effective date of an IND, and the submission date of an NDA, plus the time
between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible
for the extension and the application for extension must be made prior to expiration of the patent. The U.S. PTO, in consultation with
the FDA, reviews and approves the application for any patent term extension or restoration. In the future, Kadimastem intends to apply
for restorations of patent term for some of its currently owned patents to add patent life beyond their current expiration date, depending
on the expected length of clinical trials and other factors involved in the submission of the relevant NDA.
Market exclusivity
provisions under the FDCA can also delay the submission or the approval of certain applications. The FDCA provides a five-year period
of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical
entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety,
which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept
for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of
such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application
may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides
three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an approved NDA if new clinical investigations,
other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval
of the application, for example, for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only
the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing
the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an
applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the pre-clinical studies and adequate
and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Post-approval
Requirements
Once
an approval is granted, the FDA, European authorities and other regulatory authorities may withdraw the approval if compliance with
regulatory requirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously
unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the
market. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and
additional labeling claims, are subject to further regulatory authority review and approval. Some of these modifications, especially
adding indications, would likely require additional clinical studies. In addition, the FDA may require testing and
surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent
or limit further marketing of a product based on the results of these post-marketing programs.
Any drug product
manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things
record-keeping requirements; cGMPs; reporting of adverse experiences with the drug; providing the FDA with updated safety and efficacy
information; drug sampling and distribution requirements; notifying the FDA and gaining its approval of specified manufacturing or labeling
changes; and complying with FDA promotion and advertising requirements.
Drug manufacturers
and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with
the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and some state agencies for compliance
with cGMP and other laws.
Pursuant to the
Affordable Care Act (discussed in greater detail below), the Centers for Medicare & Medicaid Services (CMS) is required to collect
and publish information reported by applicable manufacturers about payments and other transfers of value manufacturers have made to physicians
and teaching hospitals. Such a law, when applicable to Kadimastem’s products, could increase the company’s regulatory liability
through the imposition of additional reporting and regulatory requirements. There are also an increasing number of state laws that require
manufacturers to make similar reports to states on pricing and marketing information.
Reimbursement
We face uncertainties
over the pricing of pharmaceutical products. Sales of a product candidate will depend, in part, on the extent to which the costs of a
product candidate will be covered by third-party payors, such as federal health programs, commercial insurance and managed care organizations.
These third-party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment
of healthcare costs has become a priority of federal and state governments and the prices of drugs have been a focus in this effort.
The U.S. government, state legislatures, foreign governments and third party payors have shown significant interest in implementing
cost-containment programs, including price controls, pricing transparency disclosure obligations, restrictions on reimbursement and requirements
for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies
in jurisdictions with existing controls and measures, could further limit Kadimastem’s net revenue and results. If these third-party
payors do not consider Kadimastem’s product candidates to be cost-effective compared to other therapies, they may not cover Kadimastem’s
product candidates after approved as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow
Kadimastem to sell any other product candidate on a profitable basis.
The Medicare Modernization
Act imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries under Part D. Under
Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient
prescription drugs. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug
plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription
drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all
the drugs in each category or class. The Centers for Medicare & Medicaid Services published a final rule in 2014 implementing
the Medicare Modernization Act. Contrary to the proposed rule, which would have enabled Part D plans to offer fewer drugs, the final
rule maintained the existing six protected classes of drug categories, but stated that some of the proposals not included in the final
rule could still be finalized in the future, which would impact payor formulary and coverage decisions.
The American Recovery
and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments
for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare
Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures
will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies
for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product
or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research
demonstrating benefits in a competitor’s product could adversely affect the sales of Kadimastem’s product candidates.
If third-party
payors do not consider Kadimastem’s product candidates to be cost-effective compared to other available therapies, they may not
cover Kadimastem’s product candidates as a benefit under their plans or, if they do, the level of payment may not be sufficient
to allow Kadimastem to sell Kadimastem’s product candidates on a profitable basis.
The Affordable
Care Act, enacted in March 2010, has had a significant impact on the health care industry. Some of the key changes made to date
pursuant to the ACA include an expansion of coverage for the uninsured, the creation of insurance marketplaces and increased protection
of insureds with new benefits, rights and protections. With regard to pharmaceutical products, among other things, the ACA made major
changes to the Medicare prescription drug program, which helped reduce drug costs for seniors and increased rebates and other costs for
the pharmaceutical industry. There have been judicial and congressional challenges to the ACA. In December 2017, Congress passed
and then the President Trump signed into law tax reform legislation that made significant changes to the ACA including the repeal of
the “individual mandate” that was in place to strongly encourage broad participation in the health insurance markets. On
December 14, 2018, a federal district court in Texas ruled that the ACA is unconstitutional as a result of the Tax Cuts and Jobs
Act, the federal income tax reform legislation previously passed by Congress and signed by President Trump on December 22, 2017,
that eliminated the individual mandate portion of the PPACA. The case, Texas, et al, v. United States of America, et al., (N.D. Texas),
is an outlier, but in 2019, the Fifth Circuit Court of Appeals subsequently upheld the lower court decision which was then appealed to
the United States Supreme Court. Further, in June 2021, the U.S. Supreme Court dismissed a lawsuit challenging the constitutionality
of the ACA after finding that the plaintiffs do not have standing to bring the litigation. Litigation and legislation over the ACA are
likely to continue, with unpredictable and uncertain results. Given these changes and other statements of political leaders, we cannot
predict the ultimate impact on the ACA and the subsequent effect on the pharmaceutical industry at this time. In November 2024,
Donald Trump was elected President and the Republican Party obtained control of the Senate. It is possible that additional executive
and regulatory initiatives, as well as legislation, will be pursued to enhance or reform ACA. We are not able to state with certainty
what the impact of potential legislation will be on Kadimastem’s business.
In addition, in
some non-U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements
governing drug pricing vary widely from country to country. For example, the EU provides options for its member states to restrict the
range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal
products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct
or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that
any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing
arrangements for other product candidate. Historically, products launched in the EU do not follow price structures of the United States
and generally tend to be significantly lower.
Healthcare Fraud
and Abuse Laws
In the U.S., the
research, development, testing, manufacturing, handling, storage, distribution, sale and promotion of drug products and medical devices
are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for
Medicare & Medicaid Services, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of
Inspector General), the U.S. Department of Justice, state Attorneys General, and other state and local government agencies. For
example, sales, marketing and scientific/educational grant programs must comply with the fraud and abuse provisions applicable to pharmaceutical
manufacturers, including the federal “Anti-Kickback Statute”, the Civil Monetary Penalty Statute, the Stark Law, the federal
False Claims Act, as amended, state and federal “Physician Payment Sunshine Act” laws and regulations, the privacy regulations
promulgated under the Health Insurance Portability and Accountability Act, or HIPAA, and similar state laws. Pricing and rebate programs
must comply with the Medicaid Drug Rebate Program requirements of the Omnibus Budget Reconciliation Act of 1990, as amended,
and the Veterans Health Care Act of 1992, as amended. If products are made available to authorized users of the Federal Supply
Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially
subject to federal and state consumer protection and unfair competition laws. Some of these health care laws include:
The Anti- Kickback
Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and
willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase,
order, or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid.
The federal False
Claims Act prohibits anyone from knowingly presenting, conspiring to present, making a false statement in order to present, or causing
to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services, including drugs, that
are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services.
This law also prohibits anyone from knowingly underpaying an obligation owed to a federal program. Increasingly, U.S. federal agencies
are requiring nonmonetary remedial measures, such as corporate integrity agreements in False Claims Act settlements. The U.S. Department
of Justice announced in 2016 its intent to follow the “Yates Memo,” taking a far more aggressive approach in pursuing individuals
as False Claims Act defendants in addition to the corporations.
The Physician Payment
Sunshine Act, enacted in 2010 as part of the Affordable Care Act, requires certain manufacturers of pharmaceuticals and medical devices
to annually report certain payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists
and chiropractors) and teaching hospitals, as well as investment interests held by physicians and their immediate family members. Effective
January 1, 2022, covered manufacturers will also be required to report on payments and other transfers of value to physician assistants,
nurse practitioners or clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists, and certified
nurse-midwives during the previous year. In recent years, several states in the United States have also enacted legislation
requiring pharmaceutical companies to file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing,
clinical trials and other activities, and/or register their sales representatives, as well as establish marketing compliance programs.
These laws may affect Kadimastem’s sales, marketing, and other promotional activities by imposing administrative and compliance
burdens on us. Failure to meet these requirements, to the extent they are applicable to Kadimastem’s activities, could also result
in a variety of governmental sanctions that could have a material adverse effect on Kadimastem’s business.
If Kadimastem’s
operations are found to be in violation of any of the foregoing or other applicable health care laws and regulations, we may be subject
to penalties, including significant administrative, civil and criminal penalties, monetary damages, disgorgement, imprisonment, the curtailment
or restructuring of Kadimastem’s operations, loss of eligibility to obtain approvals from the FDA, or exclusion from participation
in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid.
Other Countries
In addition to
regulations in the United States, the EU, the UK and Israel, Kadimastem is subject to a variety of other regulations governing
clinical trials and commercial sales and distribution of drugs in other countries. Whether or not a product candidate receive approval
from the FDA, approval of such product candidates must be obtained by the comparable regulatory authorities of countries other than the
United States before we can commence clinical trials or marketing of the product in those countries. The approval process varies
from jurisdiction to jurisdiction, and the time may be longer or shorter than that required for FDA approval. The requirements governing
the conduct of clinical trials and product licensing vary greatly from country to country.
The requirements
that Kadimastem must satisfy to obtain regulatory approval by government agencies in other countries prior to commercialization of any
other product candidate in such countries can be rigorous, costly and uncertain. In the European countries, UK, Canada and Australia,
regulatory requirements and approval processes are similar in principle to those in the United States. Additionally, depending on
the type of drug for which approval is sought, there are currently two potential tracks for marketing approval in the European countries:
mutual recognition and the centralized procedure. These review mechanisms may ultimately lead to approval in all EU countries, but each
method grants all participating countries some decision-making authority in product approval. The UK has a separate review period but
for a transitional period until 31 December 2022, may rely on approvals under the EU mutual recognition and/or centralized procedure.
Foreign governments also have stringent post-approval requirements including those relating to manufacture, labeling, reporting, record
keeping and marketing. Failure to substantially comply with these on-going requirements could lead to government action against the product,
us and/or Kadimastem’s representatives.
Company Information
We were established
as an Israeli company on October 6, 2008. We began trading on the TASE in June 2013.
C. Organizational
Structure
We
are a Swiss stock corporation organized under the laws of Switzerland. Our Swiss enterprise identification number is CHE-447.067.367.
Our registered and principal executive offices are located at The Circle 6, Postfach, 8058 Zurich, Switzerland, our general telephone
number is (41) 44 512 21 50 and our internet address is www.nlspharma.com.
In
April 2021, we formed a wholly owned subsidiary, NLS Pharmaceutics Inc., a Delaware corporation. In November, 2024 we also formed Merger
Sub, our a wholly owned subsidiary, in connection with the Merger. We did not have any other subsidiaries as of December 31, 2024.
D. Property,
Plant and Equipment
Our
principal executive office is located at The Circle 6, Postfach, 8058 Zurich, Switzerland, and consists of approximately 25 square meters
(approximately 270 square feet) of shared office space under an indefinite term lease that may be terminated by either party with one-month
prior notice.
We
consider that our current office space is sufficient to meet our anticipated needs for the foreseeable future and is suitable for the
conduct of our business; provided, however, that we may require additional space and facilities as our business expands.
ITEM 4A. UNRESOLVED
STAFF COMMENTS
None.
ITEM 5. OPERATING
AND FINANCIAL REVIEW AND PROSPECTS
The
following discussion and analysis should be read in conjunction with our financial statements and related notes included elsewhere in
this annual report. This discussion and other parts of this annual report contain forward-looking statements based upon current expectations
that involve risks and uncertainties. Our actual results and the timing of selected events could differ materially from those anticipated
in these forward-looking statements as a result of several factors, including those set forth under Item 3.D. “Risk Factors”
and elsewhere in this annual report. This discussion does not give effect to the possible impacts of the Merger.
Overview
We
are an emerging biopharmaceutical company engaged in the discovery and development of life-improving drug therapies to treat rare and
complex CNS disorders who have unmet medical needs.
Our
lead compound mazindol, a triple monoamine reuptake inhibitor and partial orexin receptor 2 agonist, in a proprietary ER formulation,
is being developed for the treatment of narcolepsy (lead indication) and ADHD (follow-on indication). We believe that this dual mechanism
of action will also enable Mazindol ER to provide potential therapeutic benefits in other rare and complex CNS disorders. CNS disorders
are a diverse group of conditions that include neurological, psychiatric, and substance use disorders. Recently, we launched a preclinical
program evaluating Mazindol ER as a treatment for fentanyl dependence, aiming to offer a non-opioid alternative in combating the opioid
crisis.
Our
Dual Orexin Receptor Agonist, or DOXA, platform has made significant strides. The development of AEX-41 and AEX-2 compounds showcases
our dedication to addressing unmet needs in sleep-wake disorders. Preliminary studies have yielded promising results, reinforcing our
confidence in these compounds’ potential to transform patient care.
We have no products approved
for commercialization and have never generated any revenue from product sales. Biopharmaceutical product development is a highly speculative
undertaking and involves a substantial degree of risk. It may be several years, if ever, before we complete pivotal clinical studies and
have a product candidate approved for commercialization and we begin to generate revenues and royalties from product sales. We have also
incurred significant operating losses. As of December 31, 2024, we have an accumulated deficit of $74.4 million.
As
of December 31, 2024, our cash and cash equivalents were $1.7 million. We believe that our existing cash and cash equivalents will not
be sufficient to fund our projected operating requirements for a period of one year from the issuance of the financial statements included
elsewhere in this annual report. This raises substantial doubt about our ability to continue as a going concern.
In
November 2024, we announced the Merger Agreement with Kadimastem, a clinical-stage cell therapy company specializing in “off-the-shelf”
allogeneic cell products for neurodegenerative diseases and diabetes. The Merger aims to create a Nasdaq-traded biotechnology company
with a robust portfolio of advanced therapies. Both companies’ boards of directors have unanimously approved the transaction, with
Kadimastem’s shareholders voting in favor and NLS major shareholders holding approximately 40% of NLSs’ common shares having
signed support letters for the NLS shareholders’ meeting anticipated by us to be convened soon. We currently anticipate closing
the Merger in the second quarter of 2025, pending effectiveness of NLSs’ pending registration statement filed with the SEC, Nasdaq
approval, NLSs’ shareholders’ approval and other customary closing conditions.
Components of
Operating Results
Licensing
Agreement
In
February 2019, we entered into the EF License Agreement to develop and commercialize our product candidate, Nolazol, in Latin American
countries with Eurofarma. The EF License Agreement covered the grant of non-transferable licenses, without the right to sublicense, to
Eurofarma to develop and commercialize Nolazol in Latin America. The EF License Agreement also specified our obligation to advance development
activities with respect to Nolazol in the United States.
Under
the EF License Agreement, we received a non-refundable, upfront payment, of $2.5 million in 2019 and were eligible to receive non-refundable
milestone payments of up to $16 million, based on the achievement of milestones related to regulatory filings, regulatory approvals and
the commercialization of Nolazol, as well as tiered royalty payments. As of December 31, 2023, we had long-term deferred revenues of
$2.5 million, which was going to be recognized when the development services of Nolazol were completed, and the product candidate received
applicable regulatory approval in Latin America that would allow Eurofarma to commence commercialization of Nolazol in accordance with
the EF License Agreement.
On
August 28, 2024, we agreed with Eurofarma to terminate the EF License Agreement effective September 30, 2024. Neither party has any claims
against the other in relation to the EF License Agreement and its termination. As of December 31, 2024, we recognized $2.5 million from
the EF License Agreement in the Statements of Operations and Comprehensive loss as Other income due to the termination of the EF License
Agreement.
Recent Financing
Agreements
Private Placements
On
September 28, 2023, we entered into a short term loan agreement, or the Short Term Loan Agreement, with Ronald Hafner, the Company’s
Chairman of the Board of Directors, or the Short Term Lender, providing for an unsecured loan to the Company in the aggregate amount
of CHF 500,000, or the Short Term Loan. Pursuant to the Short Term Loan Agreement, the Short Term Loan bears interest at a rate of 10%
per annum and matures on November 30, 2023. On November 15, 2023, the Company, entered into a series of short term loan agreements, or
the Bridge Loan Agreements and together with the Short Term Loan Agreement, the Loan Agreements, with certain existing shareholders of
the Company, including Ronald Hafner, the Company’s Chairman of the Board of Directors, Felix Grisard, Jürgen Bauer and Maria
Nayvalt, or the Bridge Lenders and together with the Short Term Lender, the Lenders, providing for an unsecured loan to the Company in
the aggregate amount of CHF 875,000.00 (approximately $1,000,000.00), or the Bridge Loan. Pursuant to the Bridge Loan Agreements, the
Bridge Loans bear interest at a rate of 10% per annum and mature on the earlier of June 30, 2024 or a liquidity event with a strategic
partner.
On
March 18, 2024, we entered into an addendum to the Short Term Loan Agreement with the Short Term Lender, and a series of addendums to
the Bridge Loan Agreements with the Bridge Lenders, each providing for an extension of the maturity date under the Loan Agreements to
December 31, 2024.
On
May 13, 2024, we entered into the second short term addendum, or the Second Short Term Addendum, to the Short Term Loan Agreement dated
September 28, 2023, and a short term addendum, or the Short Term Addendum, to the Short Term Loan Agreement dated November 15, 2023,
with the Short Term Lender providing for an extension of the maturity date under the Short Term Loan Agreements to June 30, 2025.
On
March 20, 2024, we entered into a securities purchase agreement, or the March Purchase Agreement, providing for the issuance in a registered
direct offering of 7,000,000 common shares, at a purchase price of $0.25 per share. The offering closed on March 22, 2024. In addition,
pursuant to the March Purchase Agreement, the investors received unregistered warrants, or the Common Warrants, to purchase up to an
aggregate of 3,500,000 common shares at an exercise of $0.25 per share in a concurrent private placement. The Common Warrants were immediately
exercisable upon issuance and will expire five years following the date of issuance. The March Purchase Agreement contains customary
representations and warranties and agreements of the Company and the investors and customary indemnification rights and obligations of
the parties. Pursuant to the March Purchase Agreement, we agreed not to enter into any agreement to issue or announce the issuance or
proposed issuance of any common shares or Common Share equivalents for a period of 45 days following the closing of the offering, subject
to certain customary exceptions. We have also agreed that from the date of the March Purchase Agreement until one year after the closing
date of the offering, we shall not enter into an agreement to effect any issuance by us or any of our subsidiaries of common shares or
Common Share equivalents (or a combination of units thereof) involving a variable rate transaction. The offering resulted in gross proceeds
to us of $1,750,000. We intend to use the net proceeds from the offering for working capital and general corporate purposes.
On
June 28, 2024, we entered into a securities purchase agreement providing for the issuance in a registered direct offering of 81,944
common shares at a purchase price of $9.60 per share. In addition, the investors in the offering received unregistered warrants to purchase
up to an aggregate of 81,944 common shares at an exercise of $9.60 per share in a concurrent private placement. The common warrants were
immediately exercisable upon issuance and expire five years following the date of issuance. The agreement includes customary representations,
warranties, and indemnification provisions. We agreed not to issue or announce the issuance of any common shares or equivalents for 15 days
post-closing, with certain exceptions, and not to engage in variable rate transactions for one-year post-closing. The offering generated
gross proceeds of approximately $786,660.
On September 16, 2024,
we entered into a warrant amendment agreement with an institutional investor to amend warrants to purchase up to 172,836 common shares,
adjusting the definition of a “Fundamental Transaction” and the exercise price to CHF 0.80. In exchange for the September
Warrant Amendment, the Company agreed to adjust the exercise price in the Common Warrants to CHF 0.02, and following the Company increasing
its authorized Common Shares, issued to the investor, pre-funded warrants to purchase up to 191,431 shares of Common Shares, or the Pre-Funded
Warrants.
On
October 9, 2024, we entered into a securities purchase agreement with certain accredited investors. Under this agreement, we issued and
sold 806,452 common shares and warrants to purchase an additional 806,452 common shares, at a combined purchase price of $3.97, for aggregate
gross proceeds of $3.2 million. The warrants have a term of five years and an exercise price of $4.25 per share. Investors were granted
the right to participate in up to 50% of future offerings for one year following the closing. We also agreed not to enter into an equity
line of credit or similar agreement without the consent of the majority of the preferred shareholders. The transaction closed on October
10, 2024.
Also
on October 9, 2024, we entered into a securities purchase agreement with an accredited investor to satisfy $4.0 million of our debt by
issuing 806,452 newly designated convertible preferred shares at a purchase price of $4.96 per share. The preferred shares have a conversion
price of $4.96 per share. The investor was granted the right to purchase up to an additional $10.0 million worth of convertible preferred
shares starting six months after the closing and continuing as long as they own preferred shares. The investor also has the right to
participate in up to 50% of future offerings for one year following the closing. We agreed not to enter into an equity line of credit
or similar agreement without the consent of the majority of the preferred shareholders. This transaction also closed on October 10, 2024.
On
October 9, 2024, the Company and certain existing warrant holders entered into warrant amendment agreements, or collectively, the October
Warrant Amendment, to amend those warrants issued by Company to such holders, collectively, to purchase up to 105,843 common shares issued
to such holders, or the Existing Warrants. The October Warrant Amendment makes certain adjustment to the definition of a “Fundamental
Transaction” in Section 3(e) of the Existing Warrants. In exchange for the Amendment, the Company agreed to adjust the exercise
price in the Common Warrants to CHF 0.80 and issued to the holders Pre-Funded Warrants to purchase up to 136,648 common shares, or the
October Pre-Funded Warrants. Each October Pre-Funded Warrant is exercisable for one Common Share at an exercise price of CHF 0.80 per
share. The October Pre-Funded Warrants are immediately exercisable and may be exercised at any time until all of the October Pre-Funded
Warrants are exercised in full.
On
October 10, 2024, we successfully implemented a restructuring measure by converting the claims of related party debt holders in the amount
of $2,788,650 into 493,986 common shares. This conversion was facilitated through an ordinary capital increase, providing the necessary
shares for the debt holders, and was recorded at fair market value with no gain or loss recognized in the statement of operations.
On
December 4, 2024, we entered into a securities purchase agreement with an accredited investor, providing for the issuance in a
private placement offering of 322,580 registered common shares, at a purchase price of $3.10 per common share, for aggregate gross
proceeds of up to $1 million. The offering was structured in two tranches, with the first tranche of $500,000 closing in January
2025 following shareholder approval. The proceeds from the offering were intended to support our general corporate purposes and our
proposed merger with Kadimastem. The second tranche of $500,000 may occur, at the election of the investor, within 15 days following
the Company meeting certain conditions, including the receipt of shareholder approval and the Common Shares trading for at least ten
consecutive trading days above the purchase price of $3.10, which corresponds to an approximate 15% premium. As of the date of this
annual report, the condition for the second tranche has not yet occurred. The Company uses and intends to further use the net
proceeds from the offering for working capital and general corporate purposes.
On
March 27, 2025, we entered into a securities purchase agreement, or the March 2025 SPA, with three accredited investors. Pursuant to
the terms of the March 2025 SPA, the Company agreed to issue and sell to the investors, in a private placement offering, or the March
2025 Offering, 1,212,122 Preferred Shares with a conversion price of $1.65 per share, for aggregate gross proceeds of $2 million. Pursuant
to the terms of the March 2025 SPA, the investors may purchase up to $1 million of additional Preferred Shares on identical terms as
the initial closing, subject to the Company obtaining shareholder approval which, as of the date of this annual report, has not yet been
sought. The March 2025 Offering closed on March 28, 2025.
The
initial closing of the March 2025 Offering resulted in gross proceeds to the Company of to $2 million. The Company intends to use the
net proceeds from the March 2025 Offering for working capital and general corporate purposes, including for expenses relating to the
Merger.
Further,
on March 31, 2025, the Company entered into a Common Shares Purchase Agreement with Alpha Capital Anstalt, or Alpha, or the Facility
SPA, relating to a committed equity facility. Pursuant to the Facility SPA, the Company has the right from time to time at its option
to sell to Alpha up to $25.0 million of the common shares, subject to certain conditions and limitations set forth in the Facility SPA.
Upon
the initial satisfaction of the conditions to Alpha’s obligation to purchase common shares set forth in the Facility SPA, or the
Commencement, including that a registration statement registering the resale by Alpha of the common shares under the Securities Act that
may be sold to it by the Company under the Facility SPA, or the Initial Resale Registration Statement, is declared effective by the SEC
and a final prospectus relating thereto is filed with the SEC, the Company will have the right, but not the obligation, from time to
time at its sole discretion until the first day of the month next following the 36-month period from and after Commencement, to direct
Alpha to purchase up to a specified maximum amount of common shares as set forth in the Facility SPA by delivering written notice to
Alpha prior to the commencement of trading on any trading day. The purchase price of the common shares that the Company elects to sell
to Alpha pursuant to the Facility SPA will be 95% of the volume weighted average price of the common shares during the applicable purchase
date on which the Company has timely delivered written notice to Alpha directing it to purchase common shares under the Facility SPA.
In
connection with the execution of the Facility SPA, the Company agreed to issue a pre-funded warrant, or the Pre-Funded Warrant, to purchase
$250,000 in common shares to Alpha as consideration for its irrevocable commitment to purchase the common shares upon the terms and subject
to the satisfaction of the conditions set forth in the Facility SPA.
A. Operating
Results
Operating Expenses
Our
current operating expenses consist of two components - research and development expenses and general and administrative expenses.
Research and Development Expenses,
net
Our
research and development expenses are expensed as incurred and consist primarily of costs of third-party clinical consultants who conduct
clinical and pre-clinical trials on our behalf and expenses related to lab supplies, materials and facility costs.
Clinical
trial costs are a major component of research and development expenses. We accrue and expense clinical trial activities performed by
third parties based upon actual work completed in accordance with agreements established with clinical research organizations and clinical
sites. We determine the actual costs through monitoring patient enrollment and discussions with internal personnel and external service
providers as to the progress or stage of completion of trials or services and the agreed-upon fee to be paid for such services.
Our
research and development expenses have materially increased and will continue to increase in the future as we enter into the Phase 2/3
clinical development stage of our product candidates and initiate a number of new research initiatives that are complementary to our
existing and planned research initiatives and thereby recruit additional research and development employees.
General and
Administrative Expenses
General
and administrative expenses include personnel costs, expenses for outside professional services, and all other general and administrative
expenses. Personnel costs consist of salaries, cash bonuses and benefits. Outside professional services consist of legal fees (including
intellectual property and corporate matters), accounting and audit services, IT and other consulting fees.
Finance Expense
and Income
Other
expenses include exchange rate differences and financial expenses related to credit card fees.
Interest
expense relates to interest paid for our financing obligations.
Taxation
NLS
Pharmaceutics is subject to corporate Swiss federal, cantonal and communal taxation in the Canton of Zurich, Switzerland.
We
are entitled under Swiss laws to carry forward any losses incurred for a period of seven years and can offset our losses carried forward
against future taxes. As of December 31, 2024, we had tax loss carryforwards totaling $45.8 million. There is no certainty that we will
make sufficient profits to be able to utilize these tax loss carryforwards in full. As such, we have recorded a 100% valuation on these
tax loss carryforwards.
The
effective corporate income tax rate (federal, cantonal and communal) where we are domiciled is currently 10.6%.
Notwithstanding
the corporate income tax, the corporate capital is taxed at a rate of 0.1% (cantonal and communal tax only, as there is no federal tax
on capital).
Value
Added Tax, or VAT, is charged on all qualifying goods and services by VAT-registered businesses. An amount of 7.7% of the value of the
goods or services is added to all sales invoices and is payable to the Swiss tax authorities. Similarly, VAT paid on purchase invoices
is reclaimable from the Swiss tax authorities.
Results of Operations
The
numbers below have been derived from our audited financial statements included elsewhere herein. The discussion below should be read
along with these consolidated financial statements and it is qualified in its entirety by reference to them.
For
a comparison of the year ended December 31, 2023 to the year ended December 31, 2022, see our annual report on Form 20-F for the year
ended December 31, 2023.
Comparison of the Year Ended December
31, 2024 to the Year Ended December 31, 2023
| | |
For the Year Ended
December 31, | |
| | |
2024 | | |
2023 | |
| Research and development expenses | |
$ | 422,051 | | |
$ | 5,908,288 | |
| General and administrative expenses | |
| 3,214,224 | | |
| 5,898,775 | |
| Merger transaction costs | |
| 743,838 | | |
| - | |
| Operating loss | |
| (4,380,113 | ) | |
| (11,807,063 | ) |
| Other income due to the termination of EF License Agreement | |
| 2,499,969 | | |
| - | |
| Other income (expense), net | |
| 49,351 | | |
| (219,812 | ) |
| Interest expense | |
| (45,054 | ) | |
| (119,920 | ) |
| Interest on related party loans | |
| (104,963 | ) | |
| (25,233 | ) |
| Net loss | |
| (1,980,810 | ) | |
| (12,172,029 | ) |
| Deemed dividend | |
| (2,076,180 | ) | |
| - | |
| Net loss attributable to common shareholders | |
$ | (4,056,990 | ) | |
$ | (12,172,029 | ) |
Research
and Development Expenses
Research
and development activities are essential to our business and historically represented the majority of our costs incurred. Costs for certain
development activities, such as clinical trials, are recognized based on an evaluation of the progress to completion of specific tasks
using information from the clinical sites and our vendors. In addition to these arrangements, we expect that our total future research
and development costs will increase over current levels in line with strategy to progress the development of our product candidates,
as well as discovery and development of new product candidates.
The
following table summarizes our research and development expenses during the years ended December 31, 2024 and 2023:
| | |
For
the Year Ended
December 31, | |
| | |
2024 | | |
2023 | |
| | |
| | |
| |
| Pre-clinical development | |
$ | 86,624 | | |
$ | 270,233 | |
| Clinical development | |
| - | | |
| 1,704,873 | |
| Clinical manufacturing costs | |
| - | | |
| 2,640,030 | |
| License fee with Aexon | |
| 182,485 | | |
| 30,000 | |
| Subcontractors | |
| 152,941 | | |
| 1,228,804 | |
| Other | |
| - | | |
| 34,348 | |
| Total | |
$ | 422,051 | | |
$ | 5,908,288 | |
Our research and development,
or R&D, expenses totaled $422,051 for the year ended December 31, 2024, representing a decrease of $5,486,237, or 93%, compared to
$5,908,288 for the year ended December 31, 2023. The decrease was primarily due to delayed liquidity events and subsequent change in
strategy, as we reduced R&D costs and activities, terminated subcontractor and other R&D agreements. In 2024, we focused on Aexon
licensing for pre-clinical DOXA.
General and
Administrative Expenses
Our general and administrative expenses totaled $3,214,224 for the
year ended December 31, 2024, representing a decrease of $2,684,551, or 46%, compared to $5,898,775 for the year ended December 31, 2023.
This reduction was primarily attributable to decreases in costs related to professional services, employee payroll, directors’ and
officers’ insurance, information technology, marketing and communication, and travel.
Merger transaction costs
Our merger transaction costs
totaled $743,838 for the year ended December 31, 2024, compared to $0 for the year ended December 31, 2023. These costs were primarily
attributable to increase in legal counsel, audit and accounting fees related to the ongoing Merger transaction.
Operating
Loss
As
a result of the foregoing, our operating loss totaled $4,380,113 for the year ended December 31, 2024, representing a decrease of $7,426,950
or 63%, compared to $11,807,063 for the year ended December 31, 2023. Our operating loss reflects ongoing investment in R&D and clinical
trials as we advance our pipeline along with general and administrative operational costs.
Other Income
for Termination of EF License Agreement
Other income for termination
of EF License Agreement for the year ended December 31, 2024 was $2.5 million. On August 28, 2024, we agreed with Eurofarma to terminate
the EF License Agreement effective September 30, 2024 and we recognized $2.5 million from the EF License Agreement as Other income for
termination of EF License Agreement. This termination reflects a change in the Company’s strategic direction, as management has
decided to discontinue the development and commercialization of its product candidate, Nolazol, in Latin American countries through a
strategic partnership with Eurofarma, a Brazilian pharmaceutical company.
Other Income
(Expense), net
Other income (expense) consists of exchange rate differences. We recognized
other income of $49,351 for the year ended December 31, 2024, representing a decrease of $269,163, or 122%, compared to Other expense
$219,812 for the year ended December 31, 2023. The increase in Other income, net was attributable to favorable exchange rate differences.
Interest
Expense
Interest
expense was $45,054, for the year ended December 31, 2024 representing a decrease of $74,866, or 62%, compared to expense of $119,920,
for the year ended December 31, 2023. The decrease was due to less interest due to from notes payable no longer outstanding.
Interest
on Related Party Loans
Interest
on related party loans was $104,963, for the year ended December 31, 2024 representing an increase of $79,730, or 316%, compared to expense
of $25,233, for the year ended December 31, 2023. The increase was due to a series of short-term loan agreements with certain existing
shareholders of the Company that were forgiven in October 2024.
Net Loss
As
a result of the foregoing, our net loss totaled $1,980,810 for the year ended December 31, 2024, representing a decrease of $10,191,219,
or 84%, compared to $12,172,029 for the year ended December 31, 2023. As a pre-revenue emerging biopharmaceutical company, such losses
are expected and aligned with our strategy to achieve long-term value through successful product development.
Net Loss attributable to common shareholders
Our net loss attributable
to holders of our common shares for the year ended December 31, 2024 was $4,056,990 and for the year ended December 31, 2023 was $12,172,029.
The decrease resulted from a reduction in net loss, offset by a deemed dividend totaling $2,076,180 for the incremental value from the
modification of various warrants’ exercise price and the fair value of the warrants issued as consideration for executing the September
Warrant Amendment and the October Warrant Amendment.
B. Liquidity
and Capital Resources
Overview
During
the year December 31, 2024, we funded our operations with $5,252,725 from the issuance of common shares and preferred shares in private
offerings and $173,123 from the exercise of pre-funded warrants. As of December 31, 2024, we had $1,665,395 in cash and cash equivalents.
The table below summarizes
our cash flows for the years ended December 31, 2024 and 2023:
| | |
2024 | | |
2023 | |
| | |
| | |
| |
| Net cash used in operating activities | |
$ | (4,267,884 | ) | |
$ | (9,684,466 | ) |
| Net cash used in investing activities | |
| (3,917 | ) | |
| - | |
| Net cash provided by financing activities | |
$ | 5,039,516 | | |
$ | 1,633,746 | |
| Net increase (decrease) in cash and cash equivalents | |
$ | 767,715 | | |
$ | (8,050,720 | ) |
Operating
Activities
Net cash used in operating
activities was $4,267,884 during the year ended December 31, 2024 compared with net cash used in operating activities of $9,684,466 for
the year ended December 31, 2023. The change in cash used in operating activities for the year ended December 31, 2024 was due to our
reporting a net loss of $1,980,810 for the year ended December 31, 2024, compared with a net loss of $12,172,029 for the same period in
2023, driven mainly by a $7,426,950 decrease in total operating costs and a $2,499,969 increase in Other income due to the termination
of EF Licensing Agreement for the year ended December 31, 2024.
Until
we can generate significant recurring revenues, we expect to satisfy our future cash needs through debt and/or equity financings. We
cannot be certain that additional funding will be available to us on acceptable terms, if at all. This may raise substantial doubt about
our ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome
of this uncertainty. If funds are not available, we may be required to delay, reduce the scope of, or eliminate research or development
plans for, or commercialization efforts with respect to our product candidates.
Investing
Activities
For the year ended December
31, 2024, net cash used in investing activities was $3,917 related to purchases of property and equipment.
Financing
Activities
Net
cash provided by financing activities of $5,039,516 for the year ended December 31, 2024, consisted of $5,252,725 of net proceeds from
the issuance of common shares and preferred shares in private placement offerings and $173,123 of net proceeds from the exercise of pre-funded,
offset by $386,332 of equity issuance costs. Net cash provided by financing activities of $1,633,746 for the year ended December 31,
2023, consisted of $1,633,746 of net proceeds from short-term loan agreements with certain existing shareholders of the Company.
Current Outlook
During
2024, our operations have been primarily financed through the proceeds from the sale of our common shares, preferred shares and short
term loans obtained from related parties at the end of 2023. We have incurred losses and generated negative cash flows from operations
since inception in 2015. To date we have not generated revenues, and we do not expect to generate any significant revenue from the sale
of our product candidates in the near future.
We
expect to generate losses for the foreseeable future, and these losses could increase as we continue product development until we successfully
achieve regulatory approvals for our product candidates and begin to commercialize any approved products. We are subject to all the risks
pertinent to the development of new products, and we may encounter unforeseen expenses, difficulties, complications, delays and other
unknown factors that may harm our business. We expect to incur additional costs associated with operating as a public company and we
anticipate that we will need substantial additional funding in connection with our continuing operations. If we need to raise additional
capital to fund our operations and complete our ongoing and planned clinical studies, funding may not be available to us on acceptable
terms, or at all.
As
of December 31, 2024, our cash and cash equivalents was $1.7 million. Our existing cash and cash equivalents and access to existing
financing arrangements will not be sufficient to fund operations for a period of one year as of December 31, 2024. We expect to continue
to generate operating losses and negative operating cash flows for the next few years and will need additional funding to support
our planned operating activities through profitability. We are actively exploring a range of options to raise funds, including strategic
partnerships, out-licensing, or divestment of assets of NLS, and other future strategic actions. In March 2025 we have completed our
most recent private financing round, in addition to our 2024 financing rounds, debt conversion and forgiveness, vendor buy-outs, and
the ongoing Merger opportunity. Our future viability depends on our ability to extend payment terms with third-party creditors until
additional funds have been raised.
Going Concern
As of December 31, 2024, we had an accumulated
deficit of approximately $74.4 million and we incurred an operating loss for the year ended December 31, 2024, of approximately $4.4 million.
To date, we have dedicated most of our financial resources to achieve and maintain Phase 3 readiness, research and development, clinical
studies associated with its ongoing biopharmaceutical business and general and administrative expenses. We have not generated revenues
from our activities and have incurred substantial operating losses.
As of December
31, 2024, our cash and cash equivalents were $1.7 million. Our existing cash and cash equivalents and access to existing financing arrangements
will not be sufficient to fund operations for a period of one year from the issuance of these consolidated financial statements. We expect
to continue to generate operating losses and negative operating cash flows for the next few years and will need additional funding to
support its planned operating activities through profitability. We are actively exploring a range of options to raise funds, including
strategic partnerships, out-licensing, or divestment of our assets, and other future strategic actions. During October 2024, we completed
a private financing round, debt conversions and forgiveness, and various vendor buy-outs. Additionally, on November 4, 2024, we entered
into the Merger Agreement. Our future viability is dependent on our ability to raise capital for payment of our vendors and to support
on-going operations. There can be no assurance that such capital will be available within a sufficient period of time, in sufficient
amounts or on terms acceptable to us. These conditions raise substantial doubt about our ability to continue as a going concern beyond
one year from the date of issuance of our 2024 consolidated financial statements.
Since the fiscal year ended
December 31, 2024, the Company has undertaken a number of actions which have increased its shareholders’ equity, including, among
other actions, (i) the sale of securities for approximately $2.0 million in gross proceeds under the March 2025 SPA, and (ii) signing
a Facility SPA, under which the Company has the right from time to time at its option to sell to Alpha up to $25.0 million of the common
shares, subject to certain conditions and limitations set forth in the Facility SPA.As a result of these actions, the Company believes
that, as of the date of this report, it satisfies the stockholders’ equity requirement of at least $2.5 million pursuant to Nasdaq
Listing Rule 5550(b)(1) for continued listing on the Nasdaq Capital Market.
Accordingly, the
2024 consolidated financial statements have been prepared in conformity with U.S. GAAP, which contemplate our continuation as a going
concern for a period within one year from the issuance of our 2024 financial statements and the realization of assets and satisfaction
of liabilities in the normal course of business. The carrying amounts of assets and liabilities presented in these consolidated financial
statements do not necessarily purport to represent realizable or settlement values. Our 2024 consolidated financial statements do not
include any adjustment that might result from the outcome of this uncertainty.
Capital Resources
and Liquidity
In
connection with the merger transaction, we have prepared the following unaudited pro forma balance sheets to illustrate the impact of
this transaction. The following table compares our historical balance sheets as of December 31, 2024 with the pro forma capitalization
as adjusted for the implemented transaction.
| ● | On
a pro forma basis to give effect to the net proceeds to us from: |
| (i) | the sale of 161,290 common shares at
a purchase price of $3.10 per share on January 9, 2025 pursuant to December 2024 securities
purchase agreement which generated net proceeds of $475,000; |
| (ii) | the
exercise of warrants for a total of 76,847 common shares in connection with the October 9, 2024 Equity Purchase Agreement and pre-funded
warrants provided to certain institutional investors upon execution of warrant amendment agreements which generated net proceeds of $178,699; |
| (iii) | the sale
of 1,212,122 preferred shares issued in a private placement offering with three accredited
investors entered on March 27, 2025 with a conversion price of $1.65 per share which generated
net proceeds of $1,900,000; and |
| |
(iv) |
the par value of the preferred and common shares change from CHF 0.80
to CHF 0.03 per share, effective January 17, 2025. |
The
information in this table should be read in conjunction with and is qualified by reference to our consolidated financial statements
and related notes included elsewhere in this annual report.
| | |
As of December 31, 2024 | |
| | |
Actual | | |
Pro Forma | |
| ASSETS | |
| | |
| |
| Current assets: | |
| | |
| |
| Cash and cash equivalents | |
$ | 1,665,395 | | |
$ | 4,219,094 | |
| Prepaid expenses and other current assets | |
| 560,157 | | |
| 560,157 | |
| Total current assets | |
| 2,225,552 | | |
| 4,779,251 | |
| | |
| | | |
| | |
| Property and equipment | |
| 7,290 | | |
| 7,290 | |
| Other assets | |
| 283 | | |
| 283 | |
| Total assets | |
$ | 2,233,125 | | |
$ | 4,786,824 | |
| | |
| | | |
| | |
| LIABILITIES AND SHAREHOLDERS’ EQUITY | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Accounts payable | |
$ | 515,486 | | |
$ | 515,486 | |
| Other accrued liabilities | |
| 311,278 | | |
| 311,278 | |
| Total current liabilities | |
| 826,765 | | |
| 826,764 | |
| | |
| | | |
| | |
| Commitments and contingencies | |
| | | |
| | |
| | |
| | | |
| | |
| Shareholders’ equity: | |
| | | |
| | |
| Preferred shares, CHF 0.80 ($0.80) par value, 206,452 registered shares issued and outstanding at December 31, 2024 and Pro Forma CHF 0.03 ($0.03) par value, 1,418,573 registered issued and outstanding | |
| 2,740,958 | | |
| 48,648 | |
| Common shares, CHF 0.80 ($0.80) par value, 3,159,535 and 810,722 registered shares issued and outstanding at December 31, 2024 and 2023, respectively, and Pro Forma CHF 0.03 ($0.03) par value, 3,757,672 registered issued and outstanding | |
| 166,353 | | |
| 126,789 | |
| Additional paid-in capital | |
| 72,820,671 | | |
| 78,106,244 | |
| Accumulated deficit | |
| (74,430,474 | ) | |
| (74,430,474 | ) |
| Accumulated other comprehensive loss | |
| 108,853 | | |
| 108,853 | |
| Total shareholders’ equity | |
| 1,406,361 | | |
| 3,960,060 | |
| Total liabilities and shareholders’ equity | |
$ | 2,233,125 | | |
$ | 4,786,824 | |
The anticipated
transaction is expected to strengthen our balance sheet by reducing our leverage and increasing our equity base. This will enhance our
financial flexibility and support our growth initiatives.
There
can be no assurance that capital will be available within a sufficient period of time, in sufficient amounts or on terms acceptable to
us. These conditions raise substantial doubt about our ability to continue as a going concern beyond one year from the issuance of these
unaudited interim condensed financial statements.
As
of December 31, 2024, our cash and cash equivalents were $1.7 million. We believe that our existing cash and cash equivalents will not
be sufficient to fund our projected operating requirements for a period of one year from the issuance of the financial statements included
elsewhere in this annual report. This raises substantial doubt about our ability to continue as a going concern. Additionally, our operating
plans may change as a result of many factors that may currently be unknown to us including:
| |
● |
the progress and costs
of the Company’s pre-clinical studies, clinical trials and other research and development activities; |
| |
|
|
| |
● |
the scope, prioritization
and number of the Company’s clinical trials and other research and development programs; |
| |
|
|
| |
● |
any cost that the Company
may incur under in- and out-licensing arrangements relating to its product candidate that it may enter into in the future; |
| |
|
|
| |
● |
the costs and timing of
obtaining regulatory approval for the Company’s product candidates; |
| |
|
|
| |
● |
the costs of filing, prosecuting,
enforcing and defending patent claims and other intellectual property rights; |
| |
|
|
| |
● |
the costs of, and timing
for, strengthening the Company’s manufacturing agreements for production of sufficient clinical and commercial quantities of
its product candidates; |
| |
|
|
| |
● |
the potential costs of
contracting with third parties to provide marketing and distribution services for the Company or for building such capacities internally;
and |
| |
|
|
| |
● |
the costs of acquiring
or undertaking the development and commercialization efforts for additional therapeutic applications of the Company’s product
candidates and the magnitude of the Company’s general and administrative expenses. |
Off-Balance
Sheet Arrangements
We
have not engaged in any off-balance sheet arrangements, such as the use of unconsolidated subsidiaries, structured finance, special
purpose entities or variable interest entities.
Quantitative
and Qualitative Disclosure About Market Risk
We
are exposed to market risks in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial
position due to adverse changes in financial market prices and rates. Our current investment policy is to invest available cash in bank
deposits with banks that have a credit rating of at least A-. Accordingly, a substantial majority of our cash and cash equivalents is
held in deposits that bear interest. Given the current low rates of interest we receive, we will not be adversely affected if such rates
are reduced. Our market risk exposure is primarily a result of foreign currency exchange rates, which is discussed in detail in the following
paragraph.
Foreign Currency
Exchange Risk
Our
results of operations and cash flow are subject to fluctuations due to changes in foreign currency exchange rates. As discussed above,
the vast majority of our liquid assets is held in U.S. dollars, and a certain portion of our expenses are denominated in CHF or EUR.
For instance, during the year ended December 31, 2024, approximately 59.3% of our expenses were denominated in CHF, 6.4% in EUR and 0.1%
in GBP. Changes of 5.0% and 10.0% in the U.S. dollar/CHF exchange rate would have increased/decreased our operating expenses by 3.0%
and 6.0%, respectively. However, these historical figures may not be indicative of future exposure, as we expect that the percentage
of our CHF denominated expenses will materially decrease in the near future, therefore reducing our exposure to exchange rate fluctuations.
We
do not hedge our foreign currency exchange risk. In the future, we may enter into formal currency hedging transactions to decrease the
risk of financial exposure from fluctuations in the exchange rates of our principal operating currencies. These measures, however, may
not adequately protect us from the material adverse effects of such fluctuations.
JOBS Act
Accounting Election
Under
the JOBS Act, an EGC can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until
such time as those standards apply to private companies. We have irrevocably elected to avail ourselves of this exemption from new or
revised accounting standards, and, therefore, will not be subject to the same new or revised accounting standards as public companies
that are not EGCs.
C. Research
and development, patents and licenses, etc.
For
a description of our research and development programs and the amounts that we have incurred over the last two years pursuant to those
programs, please see “Item 5. Operating and Financial Review and Prospects — A. Operating Results - Operating Expenses
- Research and Development Expenses, net” and “Item 5. Operating and Financial Review and Prospects — A. Results
of Operations - Comparison of the year ended December 31, 2024, to the year ended December 31, 2023- Research and Development Expenses,
net.”
D. Trend information
We
are not aware of any material recent trends besides those discussed elsewhere in this annual report.
E. Critical
Accounting Estimates
Critical Accounting
Policies and Estimates
Our
management’s discussion and analysis of our financial condition and results of operations is based on our financial statements,
which have been prepared in accordance with generally accepted accounting principles, or U.S. GAAP. The preparation of these financial
statements requires us to make judgments and estimates that affect the reported amounts of assets and liabilities and the disclosure
of contingent assets and liabilities at the date of the financial statements, as well as the expenses incurred during the reporting periods.
Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances,
the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent
from other sources. Actual results may differ from these judgments and estimates under different assumptions or conditions and any such
differences may be material. We believe that the accounting policies discussed below are critical to understanding our historical and
future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
Revenue Recognition
Under
ASC 606, an entity recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the
consideration which the entity expects to receive in exchange for those goods or services. To determine the appropriate amount of revenue
to be recognized for arrangements determined to be within the scope of ASC 606, we perform the following five steps: (i) identification
of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations
including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint
on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when
(or as) we satisfy each performance obligation. We only apply the five-step model to contracts when it is probable that the entity will
collect consideration it is entitled to in exchange for the goods or services it transfers to the customer.
In
February 2019, we entered into the EF License Agreement to develop and commercialize our product candidate, Nolazol, in Latin American
countries with Eurofarma. The EF License Agreement covered the grant of non-transferable licenses, without the right to sublicense, to
Eurofarma to develop and commercialize Nolazol in Latin America. The EF License Agreement also specified our obligation to advance development
activities with respect to Nolazol in the United States.
Under
the EF License Agreement, we received a non-refundable, upfront payment, of $2.5 million in 2019 and were eligible to receive non-refundable
milestone payments of up to $16,000,000, based on the achievement of milestones related to regulatory filings, regulatory approvals and
the commercialization of Nolazol, as well as tiered royalty payments. As of December 31, 2023, we had long-term deferred revenues of
$2.5 million, which was going to be recognized when the development services of Nolazol were completed, and the product candidate received
applicable regulatory approval in Latin America that would allow Eurofarma to commence commercialization of Nolazol in accordance with
the EF License Agreement.
On
August 28, 2024, we agreed with Eurofarma to terminate the EF License Agreement effective September 30, 2024. Neither party has any claims
against the other in relation to the EF License Agreement and its termination. As of December 31, 2024, we recognized $2.5 million from
our exclusive EF License Agreement in the Statements of Operations and Comprehensive loss as Other income due to the termination of the
EF License Agreement.
Pension Obligations
We
have a single insurance collective pension plan that is fully insured and operated by an insurance company which covers the
employees. Both we and the participants provide monthly contributions to the pension plan that are based on the covered salary. A
portion of the pension contribution is credited to employees’ savings accounts which earns interest at the rate provided in
the plan. The pension plan provides for retirement benefits as well as benefits on long-term disability and death. The pension plan
qualifies as a defined benefit plan in accordance with U.S. GAAP. As such, the cost of the defined pension arrangement is determined
based on actuarial valuations. An actuarial valuation assumes the estimation of discount rates, estimated returns on assets, future
salary increases, mortality figures and future pension increases. Because of the long-term nature of these pension plans, the
valuation of these is subject to uncertainties. As of December 31, 2024, the Company does not have any active employees participating in a defined benefit pension plan. However, the
Company continues to maintain a capped pension obligation related to survivor benefits payable to orphans of a former employee. This obligation
is fully covered by existing plan assets and does not give rise to any unfunded liability. All other obligations and liabilities associated
with the Company’s prior pension plan have been fully settled and transferred. As a result, the Company has no net pension-related
liability or associated pension expense recognized on its balance sheet as of December 31, 2024.
Income Taxation
We
incur tax loss carryforwards generating deferred tax assets against which a valuation allowance is recorded when it is not more likely
than not that the tax benefit can be realized. Judgment is required to determine the use of tax loss carryforwards. Management’s
current judgment is that it is not more likely than not that the tax benefits can be realized and a full valuation allowance is therefore
recognized.
ITEM 6. DIRECTORS,
SENIOR MANAGEMENT AND EMPLOYEES
A. Directors
and Senior Management
The
following table sets forth information regarding our directors and senior management and directors as of May 15, 2025:
| Name |
|
Age |
|
Position |
|
Initial
Year of
Appointment |
| Directors |
|
|
|
|
|
|
| Ronald Hafner(1)(3) |
|
61 |
|
Chairman and Director |
|
2015 |
| Alexander Zwyer |
|
56 |
|
Chief Executive Officer
and Director |
|
2015 |
| Nicole Fernandez-McGovern |
|
52 |
|
Chief Financial Officer |
|
2024 |
| Eric Konofal |
|
57 |
|
Chief Scientific Officer |
|
2021 |
| Gian-Marco Rinaldi(1)(2)(3) |
|
56 |
|
Director |
|
2021 |
| Florence Allouche Aknin(3) |
|
58 |
|
Director |
|
2024 |
| Olivier Samuel(3) |
|
50 |
|
Director |
|
2024 |
| (1) |
Member of Compensation,
Nomination & Governance Committee |
| (2) |
Member of the Audit Committee |
| (3) |
Independent Director (as
defined under Nasdaq rules) |
In
general, members of our board of directors are elected by our shareholders and elected to serve for a one-year period.
Ronald Hafner,
Chairman of the Board of Directors
Ronald
Hafner has been a member of our board of directors and served as Chairman of the board of directors since August 2015. Mr. Hafner
has been a partner at Deloitte Consulting AG since January 2016 and from 2018 to 2025 he has served as the chairman of Magnetic Rock
Investments Ltd., a private investment company. From 2020 to 2023 he served as a director of Norline AG. From 2015 to 2016, Mr. Hafner
served as an independent director of the Pingar Group Ltd., a private company developing artificial intelligence software solutions.
Prior to serving in these capacities, Mr. Hafner served as the Chief Executive Officer of Infosys Consulting AG. Prior to Lodestone Management
Consultants AG’s acquisition by Infosys Ltd. in 2012, Mr. Hafner was Chief Executive Officer and partner of Lodestone Management
Consultants AG, where he was responsible for the company’s global operations since founding the business in 2005. Mr. Hafner has
an M.B.A. and master’s degree in economics from the University of Basel/Switzerland.
Alexander Zwyer,
Chief Executive Officer and Director
Alexander
Zwyer has served as NLS’s Chief Executive Officer and as a Director since NLS’s incorporation in August 2015.
Mr. Zwyer has over 25 years of international business experience. In 2007, prior to, and until founding NLS in 2015, Mr. Zwyer
founded a start-up in the high-end luxury food sector and served as its chief executive officer until 2015 when he successfully sold
the company. From 1991 and until 2007, Mr. Zwyer served in various positions with Viforpharma AG (SWX: VIFN) (then known as
Vifor (International) Inc.), most notably serving as Executive Vice President (chief operating officer), leading the company’s
global regulatory affairs, medical affairs, sales and marketing as well as business development teams. Mr. Zwyer speaks seven languages
fluently. Mr. Zwyer holds a B.B.A. in business administration from Oekral, Zurich, Switzerland and an executive M.B.A. from GSBA/Lorange
Institute of Business, Zurich, Switzerland and an M.B.A. from University at Albany SUNY.
Nicole Fernandez–McGovern,
Chief Financial Officer
Nicole
Fernandez-McGovern, has served as the Chief Financial Officer of NLS since October 2024. Ms. Fernandez-McGovern is an accomplished
financial executive with over 25 years of experience blending entrepreneurial thinking with Fortune 500 best practices in finance and
operations. Ms. Fernandez-McGovern is President of RCM Financial Consulting, offering strategic advisory services. She was Interim CFO
of Hayden AI, a leading-edge technology company providing vision AI solutions for smart cities, from October 2023 to May 2024,
contributing to $115 million in capital raises and securing a $700 million contract with New York’s Metropolitan
Transportation Authority. From 2016 to 2023, she served as CFO and EVP of Operations at AgEagle Aerial Systems (NYSE: UAVS), a NYSE-listed
manufacturer of advanced commercial drone hardware and technology SAAS solutions for the defense, commercial and public safety sectors
overseeing global manufacturing and financial operations. Ms. Fernandez-McGovern served as audit chair and board member of AGO Global,
Inc. from 2011 through 2023. Previously, she served as CEO and CFO of Trunity Holdings, Inc. from 2012 to 2016. Ms. Fernandez-McGovern
holds both a BBA and MBA from the University of Miami, has been a licensed Certified Public Accountant in Florida since 1998, serves
on multiple boards, and is fluent in Spanish.
Eric Konofal,
Chief Scientific Officer
Eric
Konofal, MD, Ph.D. has served as our Chief Scientific Officer since February 2021 and as our Interim Chief Medical Officer since
January 2024. He also served as our director from December 2015 to December 2020. Dr. Konofal is a globally recognized physician-scientist
and co-founder of NLS Pharmaceutics Ltd. He is also the President and founder of Aexon since November 2023. Dr. Konofal has been active
for more than 30 years in the field of sleep research, including narcolepsy and hypersomnia, as a clinician, scientific researcher, and
drug hunter. From 2006 to date, Dr. Konofal serves as a senior medical consultant for the Pediatric Sleep Disorders Center and the Child
and Adolescent Psychiatry Department at Robert Debré Hospital (APHP). Dr. Konofal received his medical doctorate and Ph.D. from
the University Pierre — Marie Curie, Paris/France.
Gian-Marco Rinaldi, Director
Gian-Marco
Rinaldi has served as a Director since June 2021. Mr. Rinaldi has over 25 years of international business experience. Since 2009,
Mr. Rinaldi worked at Equity Partner Finad AG, Multi Family Office as an Advisor on financial matters with focus on Private Equity. Mr.
Rinaldi has served as a director of private companies, including, Tupacta AG since 2020 and Jamahook AG, Widinvest AG and Larimed since
2021. He has also served as a director of Wandele Holding AG since 2022, a director of SPitch AG since 2023 and of Swissifico AG since
2024. From 2007 and until 2009, Mr. Rinaldi served in various positions with Clariden Leu, as a Managing Director. From 1995 and until
2000, Mr. Rinaldi served as Manager distressed assets in Corporate Banking for UBS. Mr. Rinaldi speaks three languages fluently. Mr.
Rinaldi holds a Master of Advanced Studies at University of Applied Sciences Nordwestschweiz, Switzerland and a Master in law at University
of Zurich, Switzerland.
Florence Allouche Aknin, Director
Florence
Allouche Aknin has served as a Director since January 2024. From July 2022 to October 2023, Dr. Allouche Aknin served as the Chairperson
of the board of directors of Genomic Vision S.A. and from July 2016 to October 2020 she served as the President and Chief Executive Officer
of SparingVision. Dr. Allouche Aknin is a Doctor of Pharmacy and holding a Master’s in Business Administration, began her career
as a biologist at AP-HP (Greater Paris Hospitals), managing the tech transfer office for 16 years and creating 75 start-ups. Dr. Allouche
Aknin is an expert and mentor at the Paris incubators and has represented French technology internationally. She is a Professor of Pharmacy
at the University Paris City and an elected member of the French National Academy of Pharmacy. She is also the President and Founder
of Myrpharm Advisors, a start-up studio for the biopharmaceutical industry. In addition, in 2022, Dr. Allouch Aknin launched Sorbonne
Venture, a €100M fund dedicated to health technology. She is also Chairwoman of the Board of Directors for PepKon, a French biotechnology
company. Among her many awards, Dr. Allouche Aknin was elected “Woman of the Year 2017” by the French financial magazine
La Tribune, Women’s trajectory Award 2018 by HEC Paris, and Mercure Entrepreneurs Award 2019 by HEC Paris.
Olivier Samuel,
Director
Olivier
Samuel has served as our director since October 2024. He has also served as a director of Kadimastem since December 2020. Mr. Samuel
brings over 25 years of experience in product sales in the medical space. Mr. Samuel is also a professional mediator since
March 2021. From January 1997 until December 2019, Mr. Samuel was the CEO of Adomsante in France. Mr. Samuel has
a master’s in business administration and management from Emlyon Business School and the Lille School of Health Engineers.
Olivier Samuel,
Director
Mr. Samuel
has served as director of Kadimastem since January 2020. Mr. Samuel brings over 25 years of experience in product sales
in the medical space. From March 2021 and until today, Mr. Samuel has served as a professional mediator is a self-employed
Professional Mediator. From January 1997 until December 2019, Mr. Samuel was the CEO of Adomsante in France. Mr. Samuel
has a master’s in business administration and management from Emlyon Business School and the Lille School of Health Engineers.
Family Relationships
There
are no family relationships among any of our senior management and our directors.
Arrangements
for Election of Directors and Members of Senior Management
There
are no arrangements or understandings with major shareholders, customers, suppliers or others pursuant to which any of our senior management
or our directors were selected.
B. Compensation
Compensation
The
following table presents in the aggregate all compensation we paid to all of our directors and senior management as a group for the year
ended December 31, 2024. The table does not include any amounts we paid to reimburse any of such persons for costs incurred in providing
us with services during this period. For the year 2024, pursuant to Swiss law, we are required to provide the compensation report detailing
the compensation (i) in a total amount and individual amount of our members of the board of directors (stating the name and function
of the member concerned), (ii) in a total amount and the highest amount attributable to the senior management (stating the name and function
of the member concerned) and (iii) name and function of senior management who received an additional amount (if any) to the general meeting
of shareholders.
All
amounts reported in the table below reflect the cost to the Company, in thousands, for the year ended December 31, 2024.
| |
|
Salary,
Bonuses
and
Related
Benefits |
|
|
Pension,
Retirement
and Other
Similar
Benefits |
|
|
Share
Based
Compensation |
|
| All directors and senior management as a group, consisting of 12 persons |
|
$ |
901 |
|
|
$ |
112 |
|
|
$ |
0 |
|
In
accordance with Swiss law, we are required to disclose the compensation granted to the board of directors and the senior management as
follows: the total amount for the board of directors and the amount attributable to each member of the board of directors, stating the
name and function of the member concerned, as well as the total amount for the senior management and the highest amount attributable
to a member of the senior management, stating the name and function of the member concerned. The table below reflects the compensation
granted during or with respect to the year ended December 31, 2024.
All
amounts reported in the table below reflect the cost to the Company, in thousands, for the year ended December 31, 2024:
| |
|
Salary,
Bonuses
and
Related
Benefits(6) |
|
|
Pension,
Retirement
and Other
Similar
Benefits |
|
|
Share Based
Compensation |
|
| Ronald Hafner |
|
$ |
15 |
|
|
$ |
7 |
|
|
$ |
0 |
|
| Alexander Zwyer |
|
$ |
550 |
|
|
$ |
60 |
|
|
$ |
0 |
|
| Claudio L. A. Bassetti(1) |
|
$ |
0 |
|
|
$ |
0 |
|
|
$ |
0 |
|
| George Apostol(2) |
|
$ |
5 |
|
|
$ |
1 |
|
|
$ |
0 |
|
| Gian-Marco Rinaldi |
|
$ |
10 |
|
|
$ |
4 |
|
|
$ |
0 |
|
| Audrey Greenberg(3) |
|
$ |
27 |
|
|
$ |
3 |
|
|
$ |
0 |
|
| Florence Aknin Allouche |
|
$ |
10 |
|
|
$ |
2 |
|
|
$ |
0 |
|
| Anthony Walsh(4) |
|
$ |
17 |
|
|
$ |
1 |
|
|
$ |
0 |
|
| Olivier Samuel |
|
$ |
0 |
|
|
$ |
0 |
|
|
$ |
0 |
|
| Elena Thyen-Pighin(5) |
|
$ |
192 |
|
|
$ |
29 |
|
|
$ |
0 |
|
| Nicole Fernandez-McGovern |
|
$ |
54 |
|
|
$ |
0 |
|
|
$ |
0 |
|
| Eric Konofal |
|
$ |
108 |
|
|
$ |
0 |
|
|
$ |
0 |
|
| (1) |
Resigned as a member of
the Company’s board of directors on November 15, 2024. |
| (2) |
Resigned as a member of
the Company’s board of directors on May 28, 2024. |
| (3) |
Resigned as a member of
the Company’s board of directors on April 18, 2025. |
| (4) |
Resigned as a member of
the Company’s board of directors on April 3, 2024. |
| (5) |
Terminated as Chief Financial
Officer, effective as of October 31, 2024. |
| (6) |
Includes compensation received in common shares as
follows: |
| |
● |
Mr. Ronald Hafner was granted 1,251 common shares in consideration for board fees of $7,060; |
| |
● |
Mr. Alexander Zwyer was granted 814 common shares in consideration for board fees of $4,595; |
| |
● |
Mr. George Apostol was granted 366 common shares in consideration for board fees of $2,068; |
| |
● |
Mr. Gian-Marco Rinaldi was granted 807 common shares in consideration for board fees of $4,555; |
| |
● |
Ms. Audrey Greenberg was granted 266 common shares in consideration for board fees of $1,502; and |
| |
● |
Ms. Florence Aknin Allouche was granted 266 common shares in consideration for board fees of $1,502. |
Employment
Agreements with Senior Management
We
have entered into written employment or service agreements with our Chief Executive Officer, Chief Financial Officer and Chief Scientific
Officer, currently our only members of senior management. Except for our Chief Executive Officer, our other members of the management
team are not engaged as full-time employees as of December 31, 2024. We have entered into consulting or employment agreements with our
other management and employees. All of these agreements contained provisions regarding noncompetition, confidentiality of information
and assignment of inventions. However, the enforceability of the noncompetition provisions may be limited under applicable law and, our
assignment of inventions agreements with our senior management contain terms and conditions customary under Swiss law. In addition, our
senior management are subject to our directors’ and officers’ liability insurance policy, which we obtained prior to the
initial public offering. Members of our senior management are eligible for bonuses each year. The bonuses will be payable upon meeting
objectives and targets that are set annually by our board of directors and compensation and governance committee, and, in certain circumstances,
upon approval by our shareholders.
C. Board Practices
Board of Directors
On
May 3, 2023, our board of directors resolved to consider increasing the size of the board of directors by two seats and seek U.S. based,
industry experts as candidates to potentially serve as independent directors at our next annual general shareholders meeting.
Our
board of directors is currently composed of five directors. Each director is elected for a one-year term. Messrs. Hafner, Rinaldi and
Zwyer were reappointed at the annual shareholders’ meeting held on June 30, 2023. Audrey Greenberg and Anthony Walsh were elected
to the board of directors at the annual shareholders’ meeting held on June 30, 2023. Claudio L.A. Bassetti and Florence Allouche
Aknin joined the board of directors effective January 1, 2024. Mr. Walsh resigned from the board of directors on April 3, 2024. Dr. Apostol
resigned from the board of directors on May 28, 2024. Mr. Samuel was elected to the board of directors at an extraordinary shareholders’
meeting held on October 3, 2024. Dr. Bassetti resigned from the board of directors on November 15, 2024. Ms. Greenberg resigned from
the board of directors on April 18, 2025. The resignations are consistent with the Company’s ongoing efforts to reduce cash burn
and aligns with the anticipated post-merger composition of the board of directors, pursuant to which all current directors, with one
exception, are expected to step down.
Swiss
law requires that any listed company exceeding two of the three thresholds specified in art. 727 para. 1 no. 2 of the Swiss Code of Obligations,
or the CO, in two successive financial years shall have each gender represented by at least 30% on the board of directors and 20% on
the senior management team. If a company fails to comply, it must be disclosed in a statutory compensation report, including an explanation
and a designation of measures to be taken to reconcile the failed compliance. For our board of directors, this rule will apply, subject
to meeting the thresholds required under the CO, from the business year 2026, whereas for the senior management team from the business
year 2031. The triggering thresholds are (i) a balance sheet total of CHF 20 million, (ii) sales revenue of CHF 40 million and (iii)
an average of 250 full-time employees per year.
Director Independence
Our
board has reviewed the independence of our directors, applying the Nasdaq independence standards. Based on this review, the board determined
that each of Florence Allouche Aknin, Olivier Samuel, Ronald Hafner and Gian-Marco Rinaldi are “independent” within the meaning
of the Nasdaq rules. In making this determination, our board considered the relationships that each of these non-employee directors has
with us and all other facts and circumstances our board deemed relevant in determining their independence. As required under applicable
Nasdaq rules, our independent directors meet on a regular basis as often as necessary to fulfill their responsibilities, including at
least annually in executive session without the presence of non-independent directors and senior management.
Committees of
the Board of Directors
Our
board of directors has established two standing committees, the audit committee, and the compensation, nomination and governance committee.
Audit Committee
Under
Nasdaq rules, we are required to maintain an audit committee consisting of at least three members, all of whom are independent and are
financially literate and one of whom has accounting or related financial management expertise. We have complied with this requirement
in accordance with Nasdaq Listing Rule 5615(b)(1).
Our
audit committee currently consists of Gian-Marco Rinaldi, Florence Allouche Aknin and Olivier Samuel, who meet the requirements for
financial literacy under Nasdaq rules. Mr. Rinaldi serves as chairperson of the audit committee and our board of directors has determined
that Mr. Rinaldi is an “audit committee financial expert” as defined by the SEC rules and has the requisite financial experience
as defined by the Nasdaq Listing Rules.
The
audit committee is governed by a charter that complies with Nasdaq rules. The audit committee has the responsibility to, among other
things:
| |
● |
consider and make recommendations
to the board of directors on our financial statements, review and discuss the financial statements with the senior management and
the company’s external independent auditor and present its recommendations with respect to the financial statements to the
board of directors prior to the approval of the financial statements at the general shareholders’ meeting; |
| |
● |
oversee our independent
auditor and engage and determine compensation or termination of engagement of our auditor; |
| |
● |
determine and pre-approve
the terms of certain audit and non-audit services provided by our auditor; |
| |
● |
review and monitor, if
applicable, legal matters with significant impact and findings of regulatory authorities, receive reports regarding irregularities
and legal compliance, and make recommendations to our board of directors if so required, and oversee our policies and procedures
regarding compliance to applicable financial and accounting related standards, rules and regulations; and |
| |
● |
review and assess the independent
auditor’s report, management letters and take notice of all comments of the independent auditor on accounting procedures and
systems of control and review the independent auditor’s reports with senior management. |
Compensation,
Nomination and Governance Committee
The
members of our compensation, nomination and governance committee are currently Ronald Hafner and Gian-Marco Rinaldi Diaz de la Cruz with
Ronald Hafner serving as chairperson. The compensation, nomination and governance committee assists our board of directors in overseeing
our cash compensation and equity award recommendations for our senior management along with the rationale for such recommendations, as
well as summary information regarding the aggregate compensation provided to our senior management. Swiss law requires that we adopt
a compensation committee, so in accordance with Nasdaq Listing Rule 5615(a)(3), we follow home country requirements with respect to the
compensation committee. In accordance with Swiss Law, the compensation of our board of directors and senior management must be presented
by the board of directors to our shareholders and our shareholders must vote on the proposed compensation. In addition, in accordance
with Nasdaq Listing Rule 5615(a)(3), we follow home country requirements with respect to the nomination and governance committee. With
respect to the nomination of persons to our board of directors, our home country rules do not require us to authorize a committee of
our independent directors or alternatively hold a vote consisting of solely our independent directors in order to determine which persons
shall be nominated for election by our shareholders.
As
a result, our practice varies from the requirements of Nasdaq Listing Rule 5605(d) and 5605(e) which set forth certain requirements as
to the responsibilities, composition and independence of compensation committees and the nomination of directors for election by shareholders,
respectively. See “Differences between Swiss Laws and Nasdaq Requirements.”
The
members of the compensation, nomination and governance committee are elected annually by the shareholders’ meeting for a period
until the completion of the next annual shareholders’ meeting and are eligible for re-election. Each member of the compensation,
nomination and governance committee is elected individually. If there are vacancies on the compensation, nomination and governance committee
and the number of members falls below the minimum of two, the board of directors shall appoint the missing member from among its members
for the remaining term of office.
D. Employees.
As
of May 15, 2025, we have three senior management members, of which our Chief Executive Officer is a full-time employee, while the other
members of the senior management team are part-time consultants. None of our employees is represented by labor unions or covered by collective
bargaining agreements. We believe that we maintain good relations with our employees.
All
of our employment and consulting agreements will include employees’ and consultants’ undertakings with respect to non-competition
and assignment to us of intellectual property rights developed in the course of employment and confidentiality.
E. Share Ownership.
Option Plan
On
December 14, 2021, the board of directors, adopted the Share Option Plan Regulation 2021. The purpose of the Option Plan is to retain,
attract and motivate management, employees, directors and consultants by providing them with options to purchase our common shares. The
board of directors allocated fifteen percent (15%) of our fully diluted shares to awards that may be made pursuant to the Option Plan.
For more information, see “Item 7.A. Major Shareholders” below.
F. Action to
Recover Erroneously Awarded Compensation.
Not
applicable.
ITEM 7. MAJOR
SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A. Major Shareholders
The
following table sets forth information regarding beneficial ownership of our common shares as of May 15, 2025 by:
| |
● |
each person, or group of
affiliated persons, known to us to be the beneficial owner of more than 5% of our voting securities. |
| |
● |
each of our directors and
senior management; and |
| |
● |
all of our directors and
senior management as a group. |
Except
as indicated in footnotes to this table, we believe that the shareholder named in this table has sole voting and investment power with
respect to all shares shown to be beneficially owned by it, based on information provided to us by such shareholder. The shareholder
listed below does not have any different voting rights from any of our other shareholders.
| |
|
No. of Shares
Beneficially
Owned(1) |
|
|
Percentage
Owned(1) |
|
| Executive Officers and Directors |
|
|
|
|
|
|
|
|
| Ronald Hafner(2) |
|
|
556,932 |
|
|
|
15.0 |
% |
| Alexander C. Zwyer(3) |
|
|
1,614 |
|
|
|
* |
% |
| Nicole Fernandez – McGovern |
|
|
— |
|
|
|
— |
|
| Eric Konofal(4) |
|
|
51,110 |
|
|
|
* |
% |
| Olivier Samuel |
|
|
— |
|
|
|
— |
|
| Gian-Marco Rinaldi(5) |
|
|
15,658 |
|
|
|
* |
% |
| Florence Allouche Aknin(6) |
|
|
5,421 |
|
|
|
* |
% |
| Sponsor and its affiliates as a group |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
| 5% or Greater Holders |
|
|
|
|
|
|
|
|
| League Jinn Sarl(7) |
|
|
415,826 |
|
|
|
10.9 |
% |
| Alpha Capital Anstalt(8) |
|
|
359,404 |
|
|
|
9.99 |
% |
| Revel Family(9) |
|
|
378,024 |
|
|
|
10.0 |
% |
| (1) |
Beneficial ownership is
determined in accordance with the rules of the SEC. Under these rules, a person is deemed to be a beneficial owner of a security
if that person, even if not the record owner, has or shares the underlying benefits of ownership. These benefits include the power
to direct the voting or the disposition of the securities or to receive the economic benefit of ownership of the securities. A person
also is considered to be the “beneficial owner” of securities that the person has the right to acquire within 60 days
by option or other agreement. Beneficial owners include persons who hold their securities through one or more trustees, brokers,
agents, legal representatives or other intermediaries, or through companies in which they have a “controlling interest,”
which means the direct or indirect power to direct the management and policies of the entity. The percentages shown are based on
3,597,641 common shares issued and outstanding as of May 15, 2025. |
| (2) |
Consists of an aggregate of (i) 437,919 common shares, (ii) 3,227 common
shares issuable upon exercise of options vested and exercisable within 60 days of May 15, 2025, (iii) 13,054 common shares issuable upon
exercise of common warrants exercisable within 60 days of May 15, 2025, (iv) 100,807 common shares issuable upon exercise of common warrants
exercisable within 60 days of May 15, 2025, subject to a beneficial ownership limitation of 9.99%, and (v) 1,925 common shares issuable
upon exercise of pre-funded warrants exercisable within 60 days of May 15, 2025, subject to a beneficial ownership limitation of 4.99%.
The beneficial ownership limitation restricts from exercising that portion of the warrants that would result in Mr. Ronald Hafner owning,
after exercise, a number of shares of common stock in excess of the beneficial ownership limitation. Mr. Hafner is Chairman of NLS Pharmaceutics
Ltd. |
| |
|
| (3) |
Consists of an aggregate of 1,614 common shares issuable upon exercise
of options within 60 days of May 15, 2025. |
| (4) |
Consists of an aggregate of (i) 48,528 common shares, and (ii) 2,582
common shares issuable upon exercise of options within 60 days of May 15, 2025. |
| (5) |
Consists of an aggregate of (i) 12,399 common shares, (ii) 1,614 common
shares issuable upon exercise of options within 60 days of May 15, 2025, and (iii) 1,645 common shares issuable upon exercise of common
warrants exercisable within 60 days of May 15, 2025. |
| |
|
| (6) |
Consists of (i) 3,005 common shares, and (ii) 2,416 common shares issuable
upon exercise of options within 60 days of May 15, 2025. |
| (7) |
Consists of (i) 207,913
common shares, and (ii) 207,913 common shares issuable upon exercise of common warrants exercisable within 60 days of May 15, 2025,
subject to a beneficial ownership limitation of 9.99%. The beneficial ownership limitation restricts from exercising that portion
of the warrants that would result in League Jinn Sarl owning, after exercise, a number of shares of common stock in excess of the
beneficial ownership limitation. Julien Ruggieri, the sole manager of League Jinn Sarl holds voting and dispositive power over the
securities held by League Jinn Sarl. The address of League Jinn Sarl is 4-6 Rue Du Fort Rheinsheim, L-2419, Luxemburg. |
| |
|
| (8) |
Consists of (i) 161,290
common shares, (ii) 757,576 common shares issuable upon conversion of preferred shares within 60 days of May 15, 2025, (iii)
700,337 common shares issuable upon exercise of common warrants within 60 days of May 15, 2025, and (iv) 441,452 common shares issuable
upon conversion of Preferred Participation Certificates within 60 days of May 15, 2025. The warrants are subject to a beneficial
ownership limitation of 9.99%, which such limitation restricts Alpha from exercising that portion of the warrants that would result
in Alpha and its affiliates owning, after exercise, a number of shares of common stock in excess of the beneficial ownership limitation.
As a result, the number of common shares reflected in the table owned by Alpha includes (a) any outstanding common shares held by
such shareholder, and (b) if any, the number of preferred shares and warrants convertible into common shares that the holder has
the right to acquire within 60 days of May 15, 2025 without such holder or any of such holder’s affiliates beneficially owning
more than the beneficial ownership limitation of 9.99%. The address for Alpha is Lettstrasse 32, Vaduz 9490, Liechtenstein. Nicola
Feuerstein, a Director of Alpha, holds voting and dispositive power over the securities held by Alpha. |
| |
|
| (9) |
Consists of (i) 189,012
common shares, and (ii) 189,012 common shares issuable upon exercise of common warrants exercisable within 60 days of May 15, 2025,
subject to a beneficial ownership limitation of 9.99%. Revel Family Ltd. is controlled by Michel Revel, its sole equity owner, who
holds voting and dispositive power over the securities. The address of Revel Family Ltd. is 52 HaNasi HaRishon Street, Rehovot, Israel,
7630242. |
Significant
Changes in Ownership by Major Shareholders
To our knowledge,
during the past three years, there have been decreases in their relative percentage of ownership of our major shareholders as a result
of our initial public offering and other equity issuances and an addition of a major shareholder due to multiple investments made in
the Company during 2024, except for the following:
| ● | As
of May 15, 2025, following issuance of common shares and warrants exercisable into common
shares of the Company in certain private placement transactions dated October 2024 (see
“Item 5. Operating and Financial Review and Prospects — Components of Operating
Results — Recent Financing Agreements”) to Mr. Ronald Hafner, our Chairman
and shareholder, and following an assignment of shares from another shareholder to Mr. Hafner
in exchange for the satisfaction of a debt held by Mr. Hafner, Mr. Hafner became a beneficial
owner of approximately 15.0% of our common shares, reflecting an increase from the percentage
owned as reported in our annual report on Form 20-F for the year ended December 31, 2024,
which stated that as of May 14, 2024, Mr. Hafner beneficially owned 4.8%. |
Record Holders
As
of the date of this annual report, there was a total of 28 holders of record of our common shares, of which at least 6 have, to the best
of our knowledge, a registered address in the United States.
We
are not controlled by another corporation, by any foreign government or by any natural or legal persons except as set forth herein, and
there are no arrangements known to us which would result in a change in control of the Company at a subsequent date.
B.
Related Party Transactions
Employment
Agreements
We
have entered into written agreements with our Chief Executive Officer, Chief Financial Officer and Chief Scientific Medical Officer.
Except for our Chief Executive Officer, other members of management are not engaged as full-time employees as of December 31, 2024.
All
employment agreements that we enter into contain provisions regarding noncompetition, confidentiality of information and assignment of
inventions. However, the enforceability of the noncompetition provisions may be limited under applicable law and, our assignment of inventions
agreements with our senior management will contain terms and conditions customary under Swiss law. In addition, our senior management
are subject to our directors and officers insurance policy. Members of our senior management are eligible for bonuses each year. The
bonuses will be payable upon meeting objectives and targets that are set annually by the board of directors and compensation, nomination
and governance committee, and, in certain circumstances, upon approval by our shareholders.
Alexander Zwyer
Employment Agreement
On
April 9, 2021, we entered into a new employment agreement with Mr. Zwyer, effective as of May 1, 2021, relating to his service as our
Chief Executive Officer. Pursuant to his employment agreement, we have agreed to pay Mr. Zwyer a base annual salary of CHF 410,400, and
a bonus payment, subject to the discretion of the board of directors. The employment agreement also provides that either party may terminate
the employment agreement with 12 months’ prior notice, and that Mr. Zwyer will be subject to a 12-month non-competition and non-solicitation
clauses. The employment agreement also provides for standard confidentiality provisions as well as reimbursement for certain expenses.
Upon the Closing of the Merger, Mr. Zwyer’s employment shall be terminated.
Eric Konofal
Consulting Agreement
In
February 2021, we entered into a consulting agreement with Mr. Eric Konofal, our current Chief Scientific Officer, pursuant to which
we agreed to pay Mr. Konofal a daily rate of CHF 2,000 for his services. The consulting agreement may be terminated by either party upon
30 days’ written notice or immediately by us in the event of a material breach by Mr. Konofal that cannot be cured. The consulting
agreement contains customary confidentiality provisions and provides for an 18-month non-solicitation clause as well as reimbursement
for certain expenses. We entered into a new consulting agreement starting July 1, 2021 for the continuation of Mr. Konofal’s engagement
with the Company in his current role. Upon the Closing of the Merger, Mr. Konofal’s consulting agreement shall be terminated
Nicole Fernandez-McGovern
Services Agreement
On
October 15, 2024, we entered into a consulting agreement with RCM Financial Consulting, Inc., to have Nicole Fernandez-McGovern provide
interim part-time Chief Financial Officer services for $18,000 per month. The consulting agreement may be terminated by either
party upon 30 days’ written notice or immediately by us in the event of a material breach by Ms. Fernandez-McGovern that cannot
be cured. The consulting agreement contains customary confidentiality provisions and provides for an 18-month non-solicitation clause
as well as reimbursement for certain expenses.
Shareholders
and Officer and Director Loans
Short Term Loan
Agreement
On September 28, 2023 and November 15, 2023, we entered into the Short
Term Loan Agreements with Ronald Hafner, the Company’s Chairman of the Board of Directors, or the Short Term Lender, providing for
unsecured loans to the Company in the aggregate amount of CHF 500,000 and CHF 250,000, respectively, or the Short Term Loans. Pursuant
to the Short Term Loan Agreements, the Short Term Loans bear interest at a rate of 10% per annum and matures on November 30, 2023.
As
previously reported, on March 18, 2024, we entered into an addendum with the Short Term Lender providing for an extension of the maturity
date under the Short Term Loan Agreement dated September 28, 2023 to December 31, 2024.
On
May 13, 2024, we entered into the second short term addendum, or the Second Short Term Addendum, to the Short Term Loan Agreement dated
September 28, 2023, and a short term addendum, or the Short Term Addendum, to the Short Term Loan Agreement dated November 15, 2023,
with the Short Term Lender providing for an extension of the maturity date under the Loan Agreements to June 30, 2025.
Bridge Loans
On November 15, 2023, we entered into the Bridge Loan Agreements with
certain existing shareholders of the Company, including Ronald Hafner, the Company’s Chairman of the Board of Directors, Felix Grisard,
Jürgen Bauer and Maria Nayvalt, or the Bridge Lenders and together with the Short Term Lender, the Lenders, providing for an unsecured
loan to the Company in the aggregate amount of CHF 875,000.00 (approximately $1,000,000.00), or the Bridge Loan. Pursuant to the Bridge
Loan Agreements, the Bridge Loans bear interest at a rate of 10% per annum and mature on the earlier of June 30, 2024 or a liquidity event
with a strategic partner.
Addendums to
Loan Agreements
On
March 18, 2024, we entered into an addendum to the Short Term Loan Agreement with the Short Term Lender, or the Short Term Loan Addendum,
and a series of addendums to the Bridge Loan Agreements with the Bridge Lenders, or the Bridge Loan Addendums each providing for an extension
of the maturity date under the Loan Agreements to December 31, 2024. On May 16, 2024, the Company entered into additional bridge loan
addendums, or the Second Bridge Loan Addendums, to the Bridge Loan Agreement dated November 15, 2023, with Felix Grisard, Jürgen
Bauer and Maria Nayvalt, providing for an extension of the maturity date under each of the Loan Agreements to June 30, 2025.
Aexon License
Agreement
On March 19, 2024, we entered
into an exclusive license agreement, or the License Agreement, with Aexon. Pursuant to the License Agreement, Aexon granted the Company
an exclusive, royalty-bearing license, or the License, with the right to grant sublicenses in multiple tiers according to the terms of
the License Agreement. Subject to earlier termination of the License Agreement in accordance with its terms, the term of the License Agreement
is from the effective date of the License Agreement to the latest of (i) the Company’s termination of the commercialization of one
or more pharmaceutical or therapeutic products, or any combination thereof, in the use of such compounds for narcolepsy and other neuro
degenerative disorders in the last region and country in which commercialization had actually begun, and (ii) the expiration of the last-to-expire
Valid Claim (as defined in the License Agreement) of a patent identified in the License Agreement and patents owned by Aexon as of the
date of the License Agreement, that covers such pharmaceutical or therapeutic product for the use of such compounds for narcolepsy and
other neuro degenerative disorders in the respective country or region in which it was used. Pursuant to the terms of the License Agreement,
the Company agreed to pay Aexon a royalty on a country-by-country basis of 5% to 30% depending on (i) earnings by the Company in a specified
region or country for licensed products covered by patents, (ii) whether the applicable patent has not been granted to the applicable
product at the time of commercialization of such product and (iii) whether the Company challenges the validity of a patent. As of the
date of this annual report, Alexander Zwyer owns 35% of Aexon, and Eric Konofal owns 59% of Aexon. Mr. Konofal is the founder of Aexon,
with which we have a license agreement, and also serves as the President of Aexon. Mr. Zwyer holds no board or executive position at Aexon
Labs.
We made an upfront
payment of $30,000 for the option exclusivity and $170,000 upon execution of the definitive agreement to exercise the option for the
License Agreement, of which $40,000 was paid in cash and the remaining $130,000 was paid in 23,028 common shares. In addition, Aexon
will receive 15% of all proceeds earned by us in any future sub-licensing agreements. We must also make royalty payments to
Aexon upon the occurrence of certain milestones. Such payments upon the occurrence of milestones contemplated in the License
Agreement range from $100,000 to $300,000, in addition to percentage royalty payments ranging from 5% to 15% which may decrease or
increase up to 30% if we challenge the validity of the patents under the agreement, depending on the result of such challenge.
Further, pursuant to the License Agreement, we agreed to pay Aexon a percentage of license fees, milestones and royalties received
from sublicensees, ranging between 5% and 15% which may decrease or increase up to 30% if we challenge the validity of the patents
under the agreement, depending on the result of such challenge. In January 2025 through April 2025, we paid $50,000 in consideration
for 50% of the proof of concept first milestone under the License Agreement.
Debt Conversion
During
the month of October 2024, we entered into Debt Forgiveness Agreements with certain lenders, including Alexander Zwyer, Ronald
Hafner, Eric Konofal, Florence Allouche Aknin, Audrey Greenberg and Gian-Marco Rinaldi, or the Lenders, pursuant to which the
Company issued an aggregate of 493,986 common shares, par value CHF 0.80 per share, or the Common Shares, of the Company, at a
conversion price of $5.65 (rounded) per share, in full satisfaction of an aggregate amount of CHF 2,089,945 ($2,296,643) and
$492,007 in loans and advances previously provided to the Company by the Lenders, or collectively, the Loan Agreements described
under “— Shareholders and Officer and Director Loans” above. This conversion was facilitated through an ordinary capital increase, providing the necessary shares for the debt holders, and was recorded
at fair market value with no gain or loss recognized in the statement of operations.
October
2024 Financings
On
October 9, 2024, we entered into a securities purchase agreement with certain accredited investors, including Ronald Hafner. Under this
agreement, we issued and sold 806,452 common shares and warrants to purchase an additional 806,452 common shares, at a combined purchase
price of $3.97, for aggregate gross proceeds of $3.2 million. The warrants have a term of five years and an exercise price of $4.25 per
share. Investors were granted the right to participate in up to 50% of future offerings for one year following the closing. We also agreed
not to enter into an equity line of credit or similar agreement without the consent of the majority of the preferred shareholders. The
transaction closed on October 10, 2024.
Also on October
9, 2024, we entered into a securities purchase agreement with an accredited investor to satisfy $4.0 million of our debt by issuing 806,452
newly designated convertible preferred shares at a purchase price of $4.96 per share. The preferred shares have a conversion price of
$4.96 per share. The investor was granted the right to purchase up to an additional $10.0 million worth of convertible preferred shares
starting six months after the closing and continuing as long as they own preferred shares. The investor also has the right to participate
in up to 50% of future offerings for one year following the closing. We agreed not to enter into an equity line of credit or similar
agreement without the consent of the majority of the preferred shareholders. This transaction also closed on October 10, 2024.
C. Interests
of Experts and Counsel
Not
applicable.
ITEM 8. FINANCIAL
INFORMATION.
A. Consolidated
Statements and Other Financial Information.
See
“Item 18. Financial Statements.”
Legal Proceedings
From
time to time we may become involved in legal proceedings that arise in the ordinary course of business. During the period covered by
the financial statements contained herein, we were not subject to any material legal proceedings that has had a material adverse effect
on our financial position, except as described below. No assurance can be given that future litigation will not have a material adverse
effect on our financial position. When appropriate in management’s estimation, we may record reserves in our financial statements
for pending litigation and other claims.
Cambrex Corporation
On
December 1, 2023, we received a letter from Cambrex Corporation, stating that as of December 1, 2023, we had an overdue balance for services
completed under certain proposals by and between the Company, Cambrex High Point, Inc. and Avista Pharma Solutions, Inc. in the aggregate
amount of $492,723.23. On October 10, 2024, the Company successfully settled the outstanding claim through a debt purchase agreement
with an accredited investor, in the amount of $200,013, which effectively resolved the creditor claim.
Clinilabs, Inc.
On
August 21, 2024, the Company received a letter from Dunn Lambert LLC, the law firm representing Clinilabs, stating that a complaint had
been filed in the Superior Court of New Jersey. The complaint concerns three unpaid invoices totaling $886,412.46, plus interest at a
rate of 6%. On June 1, 2023, Clinilabs entered into a start-up agreement with us. On December 4, 2023, we received five credit notes
and two invoices from Clinilabs pursuant to services performed by Clinilabs under the start-up agreement. Clinilabs demanded $793,112.46
from us for unpaid service fees. On October 10, 2024, the Company successfully settled the outstanding claim through a debt purchase
agreement with an accredited investor, in consideration of $500,222.91 and 49,475 common shares equal in value to $279,299.90, which
effectively resolved the creditor claim.
CoreRX, Inc.
On
December 11, 2023, we received a notice alleging several causes of action, including a failure to remit payment for services rendered
by CoreRX. On December 11, 2023, we and CoreRX agreed to a structured payment plan in which we agreed to pay CoreRX a total amount of
$1,007,700.50. On October 10, 2024, the Company successfully settled the outstanding claim through a debt purchase agreement with an
accredited investor, in the amount of $511,835, which effectively resolved the creditor claim.
Université
de Lausanne
On August 27, 2024,
the Company received correspondence from Université de Lausanne, initiating the official “audience de conciliation”
procedure, overseen by the ordinary civil court in Lausanne. The hearing was scheduled for October 9, 2024, at the Tribunal d’arrondissement
de Lausanne. The complaint pertains to an unpaid invoice for research services amounting to $110,179, plus interest at a rate of 5%.
At the hearing on October 9, 2024, Université de Lausanne was not open to discussing a potential settlement. The Company asserts
that the services provided did not meet the required standard of care and intends to defend its position. On 9 May 2025, the Company
filed a statement of defense and asserted a counterclaim in CHF 30,000 plus compensatory interests at a rate of 5% per annum since June
29, 2022.
Dividends
We
have never declared or paid any cash dividends on our common shares and do not anticipate paying any cash dividends in the foreseeable
future. Payment of cash dividends, if any, in the future will be at the discretion of our board of directors and will depend on then-existing
conditions, including our financial condition, operating results, contractual restrictions, capital requirements, business prospects
and other factors our board of directors may deem relevant.
Under
Swiss law, we may only pay dividends if we have sufficient distributable profits brought forward from the previous business years (“Gewinnvortrag”),
or if we have distributable reserves (“frei verfügbare Reserven”), each as evidenced by our audited stand-alone statutory
balance sheet prepared pursuant to Swiss law, and after allocations to reserves required by Swiss law and the articles of association
have been deducted. Furthermore, the shareholders may resolve to pay an interim dividend (“Zwischendividende”) if
we have sufficient distributable profits based on an interim account. The provisions applicable for dividends are also applicable for
the interim dividends.
Under
our amended and restated articles of association and in accordance with Swiss law, only our shareholders have the power to approve the
payment of any dividends.
B. Significant
Changes.
No
significant change, other than as otherwise described in this annual report, has occurred in our operations since the date of our financial
statements included in this annual report.
ITEM 9. THE
OFFER AND LISTING.
A. Offer and
Listing Details
Since
January 30, 2021, our common shares and Warrants trade on Nasdaq under the symbols “NLSP” and NLSPW”, respectively.
B. Plan of Distribution
Not
applicable.
C. Markets
Our
common shares and Warrants trade on Nasdaq.
D. Selling Shareholders
Not
applicable.
E. Dilution
Not
applicable.
F. Expenses
of the Issue
Not
applicable.
ITEM 10. ADDITIONAL
INFORMATION
A. Share Capital
Not
applicable.
B. Memorandum
and Articles of Association
A
copy of our amended and restated articles of association is attached as Exhibit 1.1 to this annual report. The information called for
by this Item is set forth in Exhibit 2(d) to this annual report and is incorporated by reference into this annual report.
C. Material
Contracts
The
following is a summary of each material contract, other than material contracts entered into in the ordinary course of business, to which
we are or have been a party, for the two years immediately preceding the date of this annual report:
| |
● |
ATM Sales Agreement, dated
March 4, 2022 by and between NLS Pharmaceutics Ltd. and Virtu Americas LLC. See Item 5. “Operating and Financial Review and
Prospects – Recent Financing Agreements” for more information about this document. |
| |
● |
Short Term Loan Agreement,
dated September 28, 2023 by and between NLS Pharmaceutics Ltd., and Ronald Hafner. See Item 5. “Operating and Financial Review
and Prospects – Recent Financing Agreements” for more information about this document. |
| |
|
|
| |
● |
Short Term Loan Agreements,
dated November 15, 2023, by and between NLS Pharmaceutics Ltd., and certain existing shareholders of the Company, including Ronald
Hafner, the Company’s Chairman of the Board of Directors, Felix Grisard, Jürgen Bauer and Maria Nayvalt. See Item 5. “Operating
and Financial Review and Prospects – Recent Financing Agreements” for more information about this document. |
| |
|
|
| |
● |
License Agreement, dated
March 19, 2024, by and between NLS Pharmaceutics Ltd. and Aexon Labs, Inc., a Delaware corporation. See Item 4. “Business Overview
– Additional Pre-Clinical Compounds” for more information about this document. |
| |
|
|
| |
● |
Securities Purchase Agreement,
dated March 20, 2024, by and between NLS Pharmaceutics Ltd., Armistice Capital Master Fund Ltd., and Lind Global Fund II LP. See
Item 5. “Operating and Financial Review and Prospects – Recent Financing Agreements” for more information about
this document. |
| |
|
|
| |
● |
Securities Purchase Agreement,
dated June 28, 2024, by and between NLS Pharmaceutics Ltd., Armistice Capital Master Fund Ltd., and Intracoastal Capital LLC. See
Item 5. “Operating and Financial Review and Prospects – Recent Financing Agreements” for more information about
this document. |
| |
|
|
| |
● |
Warrant Amendment Agreement,
dated September 16, 2024, by and between NLS Pharmaceutics Ltd. and Armistice Capital Master Fund Ltd. See Item 5. “Operating
and Financial Review and Prospects – Recent Financing Agreements” for more information about this document. |
| |
|
|
| |
● |
Securities Purchase Agreement,
dated October 9, 2024, by and among NLS Pharmaceutics Ltd., Rainforest Partners LLC, Revel Family LTD, Ronald Hafner, Jürgen
Bauer, Alpha, and League Jinn SARL. See Item 5. “Operating and Financial Review and Prospects – Recent Financing Agreements”
for more information about this document. |
| |
|
|
| |
● |
Securities Purchase Agreement,
dated October 9, 2024, by and between NLS Pharmaceutics Ltd. and Alpha. See Item 5. “Operating and Financial Review and Prospects
– Recent Financing Agreements” for more information about this document. |
| |
|
|
| |
● |
Agreement and Plan of Merger,
dated November 4, 2024, by and among NLS Pharmaceutics Ltd., NLS Pharmaceutics (Israel) Ltd., and Kadimastem Ltd. See Item 4. “Information
on the Company – Agreement and Plan of Merger with Kadimastem” for more information about this document. |
| |
● |
Securities Purchase Agreement, dated December 4, 2024, by and between NLS Pharmaceutics Ltd. and Alpha. See Item 5. “Operating and Financial Review and Prospects – Recent Financing Agreements” for more information about this document. |
| |
|
|
| |
● |
Securities Purchase Agreement, dated March 27, 2025, by and between NLS Pharmaceutics Ltd. and accredited investors. See Item 5. “Operating and Financial Review and Prospects – Recent Financing Agreements” for more information about this document. |
| |
|
|
| |
● |
Common Shares Purchase Agreement, dated March 31, 2025, by and between NLS Pharmaceutics Ltd. and Alpha. See Item 5. “Operating and Financial Review and Prospects – Recent Financing Agreements” for more information about this document. |
D. Exchange Controls
There are no Swiss governmental
laws, decrees or regulations that restrict, in a manner material to us, the export or import of capital, including any foreign exchange
controls, or that generally affect the remittance of dividends or other payments to non-residents or non-citizens of Switzerland who hold
our common shares.
E. Taxation.
Switzerland Tax Consideration
This summary of material Swiss
tax consequences is based on Swiss law and regulations and the practice of the Swiss tax administration as in effect on the date hereof,
all of which are subject to change (or subject to changes in interpretation), possibly with retroactive effect. The summary does not purport
to take into account the specific circumstances of any particular shareholder or potential investor and does not relate to persons in
the business of buying and selling common shares or other securities. The summary is not intended to be, and should not be interpreted
as, legal or tax advice to any particular potential shareholder/s, and no representation with respect to the tax consequences to any particular
shareholder/s is made.
Current and prospective shareholders
are advised to consult their own tax advisers in light of their particular circumstances as to the Swiss tax laws, regulations and regulatory
practices that could be relevant for them in connection with the acquiring, owning and selling or otherwise disposing of common shares
and receiving dividends and similar cash or in-kind distributions on common shares (including dividends on liquidation proceeds and stock
dividends) or distributions on common shares based upon a capital reduction (“Nennwertrückzahlungen”) or reserves paid
out of capital contributions (“Reserven aus Kapitaleinlagen”) and the consequences thereof under the tax laws, regulations
and regulatory practices of Switzerland.
Taxation of NLS Pharmaceutics Ltd.
NLS is a Swiss company, subject
to taxation in the Canton of Zurich as from December 10, 2021 onwards (formerly, Canton of Nidwalden). The Company is taxed at a current
effective income tax rate of approximately 19% (including direct federal as well as cantonal/communal taxes). In addition, an annual capital
tax rate of approximately 0.16% is levied on the net equity of the Company.
Based on a vote by
eligible Swiss voters in Switzerland on May 19, 2019, Switzerland reformed certain elements of its corporate tax law which impact
the taxation of NLS (including the abolition of the mixed company privilege at cantonal/communal level). These new federal
regulations went into effect as of January 1, 2020. Regarding the implementation of certain measures on the cantonal level, a
referendum was put up for a vote in the Canton of Nidwalden. Following a vote during the year ended December 31, 2020, the
referendum passed and took effect in the Canton of Nidwalden. Transitional measures were implemented for companies that were
previously under the abolished mixed company regime at cantonal/communal levels to prevent over-taxation on hidden reserves
generated under said regime. NLS applied for the special tax rate solution that provides for a separate taxation over five years of
the portion of the profit based on realization of hidden reserves and goodwill which were previously not taxed under the mixed
company regime. After the company changed its registered office, the same transitional measure has been applied for and granted in
the canton of Zurich.
Swiss Federal Withholding Tax on Dividends
and other Distributions
Dividend payments and similar
cash or in-kind distributions on the common shares (including dividends on liquidation proceeds and stock dividends) that the Company
makes to shareholders are subject to Swiss federal withholding tax (“Verrechnungssteuer”) at a rate of 35% on the gross amount
of the dividend. The Company is required to withhold the Swiss federal withholding tax from the dividend and remit it to the Swiss Federal
Tax Administration. Distributions based upon a capital reduction (“Nennwertrückzahlungen”) and reserves paid out of capital
contribution reserves (“Reserven aus Kapitaleinlagen”) are not subject to Swiss federal withholding tax.
The redemption of common shares
in the Company may under certain circumstances (in particular, if the common shares in the Company are redeemed for subsequent cancellation)
be taxed as a partial liquidation for Swiss federal withholding tax purposes, with the consequence that the difference between the repurchase
price and the nominal value of the shares (“Nennwertprinzip”) plus capital contribution reserves (“Reserven aus Kapitaleinlagen”)
is subject to Swiss federal withholding tax.
The Swiss federal withholding
tax is refundable or creditable in full to a Swiss tax resident corporate and individual shareholder as well as to a non-Swiss tax resident
corporate or individual shareholder who holds the common shares as part of a trade or business carried on in Switzerland through a permanent
establishment or fixed place of business situated for tax purposes in Switzerland, if such person is the beneficial owner of the distribution
and, in the case of a Swiss tax resident individual who holds the common shares as part of his private assets, duly reports the gross
distribution received in his individual income tax return or, in the case of a person who holds the common shares as part of a trade or
business carried on in Switzerland through a permanent establishment or fixed place of business situated for tax purposes in Switzerland,
recognizes the gross dividend distribution for tax purposes as earnings in the income statements and reports the annual profit in the
Swiss income tax return.
If a shareholder who is not
a Swiss resident for tax purposes and does not hold the common shares in connection with the conduct of a trade or business in Switzerland
through a permanent establishment or fixed place of business situated, for tax purposes in Switzerland, receives a distribution from the
Company, the shareholder may be entitled to a full or partial refund or credit of Swiss federal withholding tax incurred on a taxable
distribution if the country in which such shareholder is resident for tax purposes has entered into a treaty for the avoidance of double
taxation with Switzerland and the further prerequisites of the treaty for a refund have been met. Shareholders not resident in Switzerland
should be aware that the procedures for claiming treaty benefits (and the time required for obtaining a refund or credit) may differ from
country to country.
Individual and Corporate Income Tax on Dividends
Swiss resident individuals
holding the common shares as part of their private assets who receive dividends and similar distributions (including stock dividends and
liquidation proceeds), which are not repayments of the nominal value (“Nennwertrückzahlungen”) of the common shares or
reserves paid out of capital contributions (“Reserven aus Kapitaleinlagen”) are required to report such payments in their
individual income tax returns and are liable to Swiss federal, cantonal and communal income taxes on any net taxable income for the relevant
tax period. Furthermore, for the purpose of the Direct Federal Tax, dividends, shares in profits, liquidation proceeds and pecuniary benefits
from shares (including bonus shares) are included in the tax base for only 70% of their value (“Teilbesteuerung”), if the
investment amounts to at least 10% of nominal share capital of the Company. All Swiss cantons have introduced partial taxation measures
at cantonal and communal levels.
Swiss resident
individuals as well as non-Swiss resident individual taxpayers holding the common shares in connection with the conduct of a trade
or business in Switzerland through a permanent establishment or fixed place of business situated, for tax purposes, in Switzerland,
are required to recognize dividends, distributions based upon a capital reduction (“Nennwertrückzahlungen”) and
reserves paid out of capital contributions (“Reserven aus Kapitaleinlagen’) in their income statements for the relevant
tax period and are liable to Swiss federal, cantonal and communal individual or corporate income taxes, as the case may be, on any
net taxable earnings accumulated (including the payment of dividends) for such period. Furthermore, for the purpose of the Direct
Federal Tax, dividends, shares in profits, liquidation proceeds and pecuniary benefits from shares (including bonus shares) are
included in the tax base for only 70% (“Teilbesteuerung”), if the investment is held in connection with the conduct of a
trade or business or qualifies as an opted business asset (“gewillkürtes Geschäftsvermögen”) according to
Swiss tax law and amounts to at least 10% of nominal share capital of the Company. All cantons have introduced partial taxation
measures at cantonal and communal levels.
Swiss resident corporate taxpayers
as well as non-Swiss resident corporate taxpayers holding the common shares in connection with the conduct of a trade or business through
a permanent establishment or fixed place of business situated, for tax purposes, in Switzerland, are required to recognize dividends,
distributions based upon a capital reduction (“Nennwertrückzahlungen”) and reserves paid out of capital contributions
(“Reserven aus Kapitaleinlagen”) in their income statements for the relevant tax period and are liable to Swiss federal, cantonal
and communal corporate income taxes on any net taxable earnings accumulated for such period. Swiss resident corporate taxpayers as well
as non-Swiss resident corporate taxpayers holding the common shares in connection with the conduct of a trade or business through a permanent
establishment or fixed place of business situated, for tax purposes, in Switzerland may be eligible for participation relief (“Beteiligungsabzug”)
in respect of dividends and distributions based upon a capital reduction (“Nennwertrückzahlungen”) and reserves paid
out of capital contributions (“Reserven aus Kapitaleinlagen”) if the common shares held by them as part of a Swiss business
have an aggregate market value of at least CHF 1 million or represent at least 10% of the nominal share capital of the Company or give
entitlement to at least 10% of the profits and reserves of the Company, respectively.
Recipients of dividends and
similar distributions on the common shares (including stock dividends and liquidation proceeds) who neither are residents of Switzerland
nor during the current taxation year have engaged in a trade or business in Switzerland and who are not subject to taxation in Switzerland
for any other reason are not subject to Swiss federal, cantonal or communal individual or corporate income taxes in respect of dividend
payments and similar distributions because of the mere holding of the common shares.
Wealth and Annual Capital Tax on Holding of
Common Shares or Warrants
Swiss resident individuals
and non-Swiss resident individuals holding the common shares and/or Warrants in connection with the conduct of a trade or business in
Switzerland through a permanent establishment or fixed place of business situated, for tax purposes, in Switzerland, are required to report
their common shares as part of their wealth and will be subject to cantonal and communal wealth tax to the extent the aggregate taxable
net wealth is allocable to Switzerland.
Swiss resident corporate taxpayers
and non-Swiss resident corporate taxpayers holding the common shares and/or Warrants in connection with the conduct of a trade or business
in Switzerland through a permanent establishment or fixed place of business situated, for tax purposes, in Switzerland, will be subject
to cantonal and communal annual capital tax on the taxable capital to the extent the aggregate taxable capital is allocable to Switzerland.
Individuals and corporate
taxpayers not resident in Switzerland for tax purposes and not holding the common shares and/or Warrants in connection with the conduct
of a trade or business in Switzerland through a permanent establishment or fixed place of business situated, for tax purposes, in Switzerland,
are not subject to wealth or annual capital tax in Switzerland because of the mere holding of the common shares and/or Warrants.
Capital Gains on Disposal of Common Shares
or Warrants
Swiss resident individuals
who sell or otherwise dispose of the common shares and/or Warrants realize a tax-free capital gain, or a non-tax deductible capital loss,
as the case may be, provided that they hold the common shares, as part of their private assets. Under certain circumstances, the sale
proceeds may be requalified into taxable investment income (e.g., if the taxpayer is deemed to be a professional securities dealer).
Capital gains realized on
the sale of the common shares and/or Warrants held by Swiss resident individuals, as well as non-Swiss resident individuals and corporate
taxpayers holding the common shares and/or Warrants in connection with the conduct of a trade or business in Switzerland through a permanent
establishment or fixed place of business situated, for tax purposes, in Switzerland, will be subject to Swiss federal, cantonal and communal
individual tax, as the case may be. This also applies to Swiss resident individuals who, for individual income tax purposes, are deemed
to be professional securities dealers for reasons of, inter alia, frequent dealing and debt-financed purchases. Capital gains realized
by resident individuals who hold the common shares as business assets might be entitled to reductions or partial taxations similar to
those mentioned above for dividends (“Teilbesteuerung”) if certain conditions are met (e.g., holding period of at least one
year and participation of at least 10% of nominal share capital of the Company).
Swiss resident corporate taxpayers
as well as non-Swiss resident corporate taxpayers holding the common shares and/or Warrants in connection with the conduct of a trade
or business, through a permanent establishment or fixed place of business situated, for tax purposes, in Switzerland, are required to
recognize such capital gain in their income statements for the relevant tax period. Corporate taxpayers may qualify for participation
relief on capital gains (“Beteiligungsabzug”), if the common shares sold during the tax period represent at least 10% of the
Company’s share capital or if the common shares sold give entitlement to at least 10% of the Company’s profits and reserves
and were held for at least one year. The tax relief applies to the difference between the sale proceeds of common shares by the Company
and the acquisition costs of the participation (“Gestehungskosten”).
Individuals and corporations
not resident in Switzerland for tax purposes and not holding the common shares and/or Warrants in connection with the conduct of a trade
or business in Switzerland through a permanent establishment or fixed place of business situated for tax purposes in Switzerland, are
not subject to Swiss federal, cantonal and communal individual income or corporate income tax, as the case may be, on capital gains realized
on the sale of the common shares and/or Warrants.
Gift and Inheritance Tax
Transfers of common shares
and/or Warrants may be subject to cantonal and/or communal inheritance or gift taxes if the deceased or the donor or the recipient were
resident in a Canton levying such taxes.
Swiss Issuance Stamp Duty
The Company is subject to
paying to the Swiss Federal Tax Administration a 1% Swiss federal issuance stamp tax (“Emissionsabgabe”) on any increase of
the nominal share capital of the Company (including capital surplus) or any other equity contributions received by the Company (with or
without issuance of shares). Certain costs incurred in connection with the issuance of shares (if any) may be deductible. There are several
exemptions from issuance stamp tax that may apply under certain circumstances (e.g., certain intercompany reorganizations).
Swiss Securities Transfer Tax
The purchase or sale (or other
financial transfer) of the common shares, whether by Swiss residents or non-Swiss residents, may be subject to Swiss securities transfer
tax of up to 0.15%, calculated on the purchase price or the proceeds if the purchase or sale occurs through or with a bank or other securities
dealer in Switzerland as defined in the Swiss Federal Stamp Duty Act as an intermediary or party to the transaction unless an exemption
applies.
Automatic Exchange of Information in Tax Matters
On November 19, 2014, Switzerland
signed the multilateral competent authority agreement on the automatic exchange of financial account information, which is intended to
ensure the uniform implementation of automatic exchange of information, or the AEOI.
The AEOI is being introduced
in Switzerland through bilateral agreements or multilateral agreements. Switzerland has concluded a multilateral agreement with the EU
on the AEOI in tax matters, or the AEOI Agreement. This AEOI Agreement entered into force as of January 1, 2017 and applies to all 28
member states as well as Gibraltar. Furthermore, on January 1, 2017, the multilateral competent authority agreement on the automatic exchange
of financial account information and, based on such agreement, a number of bilateral AEOI agreements with other jurisdictions entered
into force. The Federal Act on the International Automatic Exchange of Information in Tax Matters, or the AEOI Act, which is the primary
legal basis for the implementation of the AEOI standard in Switzerland, entered into force on January 1, 2017 as well.
Based on such multilateral
agreements and bilateral agreements and the implementing laws of Switzerland, Switzerland collects data in respect of financial assets,
which may include Shares, held in, and income derived thereon and credited to, accounts or deposits with a paying agent in Switzerland
for the benefit of individuals resident in an EU member state or in a treaty state since 2017, and exchanges it since 2018. Switzerland
has signed and is expected to sign further bi- or multilateral AEOI in tax matter agreements with other countries, which entered into
force on January 1, 2018 or January 1, 2019 or will enter into force at a later date.
A list of such multilateral
agreements and bilateral agreements of Switzerland in effect or signed and becoming effective can be found on the website of the State
Secretariat for International Finance, or SIF.
On February 27, 2019, the
Federal Council (“Bundesrat”) initiated the consultation on the revision of the AEOI Act and AEOI Ordinance. The consultation
proposal takes account of recommendations from the Global Forum on Transparency and Exchange of Information for Tax Purposes. They concern,
among other things, certain due diligence and registration obligations, the maintenance of a document retention obligation for reporting
Swiss financial institutions, as well as definitions thereunder. Some exceptions have also been removed or adapted. The amendments to
the AEOI Act and AEOI Ordinance entered into force on January 1, 2021. Further revisions of the AEOI Act and the AEOI Ordinance concerning
the Common Reporting Standard are expected in June 2024 and to be implemented in 2026. It is expected that he amendments include a revision
of the Common Reporting Standard (CRS) for financial accounts and the addition of a new Crypto-Asset Reporting Framework (CARF).
Swiss Facilitation of the Implementation of
the U.S. Foreign Account Tax Compliance Act
Switzerland has concluded
an intergovernmental agreement with the U.S. to facilitate the implementation of the Foreign Account Tax Compliance Act, or the FATCA.
The agreement ensures that the accounts held by U.S. persons with Swiss financial institutions are disclosed to the U.S. tax authorities
either with the consent of the account holder or by means of group requests within the scope of administrative assistance. Information
will not be transferred automatically in the absence of consent, and instead will be exchanged only within the scope of administrative
assistance on the basis of the double taxation agreement between the U.S. and Switzerland. On October 8, 2014, the Swiss Federal Council
approved a mandate for negotiations with the U.S. on changing the current direct-notification-based regime to a regime where the relevant
information is sent to the Swiss Federal Tax Administration, which in turn provides the information to the U.S. tax authorities. It is
uncertain at this point when a corresponding agreement will be available.
Implementation of the OECD Global Minimum Tax in Switzerland
In light of the OECD
Base erosion and profit shifting initiative, Switzerland has introduced by means of an ordinance the OECD global minimum tax rate
for large multinational enterprises. The people and cantons approved the necessary constitutional amendment in a popular vote on
June 18, 2023. During its meeting on December 22, 2023, the Federal Council decided to implement the minimum tax rate with the
introduction of a supplementary tax in Switzerland from January 1, 2024, and in its meeting on September 4, 2024 the Federal Council
decided to additionally implement the international minimum tax from January 1, 2025. Based on such ordinance large multinational
enterprises with global turnover of at least EUR 750 million which are effectively taxed below 15% will be liable to a top-up tax
(“Ergänzungssteuer”), in line with the OECD framework for the Qualified Domestic Minimum Top-up Tax and the Income
Inclusion Rule up to 15% minimum taxation.
Material U.S. Federal Income Tax Considerations
for U.S. Holders
EACH PROSPECTIVE SHAREHOLDER SHOULD CONSULT
ITS OWN TAX ADVISOR REGARDING THE PARTICULAR SWISS TAX CONSEQUENCES OF PURCHASING, HOLDING AND DISPOSING OF OUR COMMON SHARES, INCLUDING
THE CONSEQUENCES OF ANY PROPOSED CHANGE IN APPLICABLE LAWS.
THE FOLLOWING SUMMARY IS INCLUDED
HEREIN FOR GENERAL INFORMATION AND IS NOT INTENDED TO BE, AND SHOULD NOT BE CONSIDERED TO BE, LEGAL OR TAX ADVICE. EACH U.S. HOLDER SHOULD
CONSULT WITH HIS OR HER OWN TAX ADVISOR AS TO THE PARTICULAR U.S. FEDERAL INCOME TAX CONSEQUENCES OF THE PURCHASE, OWNERSHIP AND SALE
OF COMMON SHARES, INCLUDING THE EFFECTS OF APPLICABLE STATE, LOCAL, FOREIGN OR OTHER TAX LAWS AND POSSIBLE CHANGES IN THE TAX LAWS.
Subject to the limitations
described in the next paragraph, the following discussion summarizes the material U.S. federal income tax consequences to a “U.S.
Holder” arising from the purchase, ownership and sale of the common shares. For this purpose, a “U.S. Holder” is a holder
of common shares or Warrants that is: (1) an individual citizen or resident of the United States, including an alien individual who is
a lawful permanent resident of the United States or meets the substantial presence residency test under U.S. federal income tax laws;
(2) a corporation (or entity treated as a corporation for U.S. federal income tax purposes) or a partnership (other than a partnership
that is not treated as a U.S. person under any applicable U.S. Treasury regulations) created or organized under the laws of the United
States or the District of Columbia or any political subdivision thereof; (3) an estate, the income of which is includable in gross income
for U.S. federal income tax purposes regardless of source; (4) a trust if a court within the United States is able to exercise primary
supervision over the administration of the trust and one or more U.S. persons have authority to control all substantial decisions of the
trust; or (5) a trust that has a valid election in effect to be treated as a U.S. person to the extent provided in U.S. Treasury regulations.
This summary is for general
information purposes only and does not purport to be a comprehensive description of all of the U.S. federal income tax considerations
that may be relevant to a decision to purchase our common shares or Warrants. This summary generally considers only U.S. Holders that
will own our common shares or Warrants as capital assets. Except to the limited extent discussed below, this summary does not consider
the U.S. federal tax consequences to a person that is not a U.S. Holder, nor does it describe the rules applicable to determine a taxpayer’s
status as a U.S. Holder. This summary is based on the provisions of the Internal Revenue Code of 1986, as amended, or the Code, final,
temporary and proposed U.S. Treasury regulations promulgated thereunder, administrative and judicial interpretations thereof, (including
with respect to the TCJA), and the U.S./Switzerland Income Tax Treaty, all as in effect as of the date hereof and all of which are subject
to change, possibly on a retroactive basis, and all of which are open to differing interpretations. We will not seek a ruling from the
IRS with regard to the U.S. federal income tax treatment of an investment in our common shares by U.S. Holders and, therefore, can provide
no assurances that the IRS will agree with the conclusions set forth below.
This discussion does not
address all of the aspects of U.S. federal income taxation that may be relevant to a particular U.S. holder based on such
holder’s particular circumstances and in particular does not discuss any estate, gift, generation-skipping, transfer, state,
local, excise or foreign tax considerations. In addition, this discussion does not address the U.S. federal income tax treatment of
a U.S. Holder who is: (1) a bank, life insurance company, regulated investment company, or other financial institution or
“financial services entity;” (2) a broker or dealer in securities or foreign currency; (3) a person who acquired our
common shares or Warrants in connection with employment or other performance of services; (4) a U.S. Holder that is subject to the
U.S. alternative minimum tax; (5) a U.S. Holder that holds our common shares or Warrants as a hedge or as part of a hedging,
straddle, conversion or constructive sale transaction or other risk-reduction transaction for U.S. federal income tax purposes; (6)
a tax-exempt entity; (7) real estate investment trusts or grantor trusts; (8) a U.S. Holder that expatriates out of the United
States or a former long-term resident of the United States; or (9) a person having a functional currency other than the U.S. dollar.
This discussion does not address the U.S. federal income tax treatment of a U.S. Holder that owns, directly or constructively, at
any time, common shares representing 10% or more of our voting power. Additionally, the U.S. federal income tax treatment of
partnerships (or other pass-through entities) or persons who hold common shares through a partnership or other pass-through entity
are not addressed.
Each prospective investor
is advised to consult his or her own tax adviser for the specific tax consequences to that investor of purchasing, holding or disposing
of our common shares, including the effects of applicable state, local, foreign or other tax laws and possible changes in the tax laws.
Exercise or Expiry of Warrants
No gain or loss will be realized
on the exercise of a Warrant. When a Warrant is exercised, the U.S. Holder’s cost of the common share acquired thereby will be equal
to the U.S. Holder’s adjusted cost basis of the Warrant plus the exercise price paid for the common share. The expiry of an unexercised
Warrant will generally give rise to a capital loss equal to the adjusted cost basis to the U.S. Holder of the expired Warrant. The holding
period of the common share acquired through the exercise of a Warrant includes the holding period of the Warrant. See “Passive Foreign
Investment Companies,” for the impact of the exercise of a Warrant on taxation of a U.S. Holder if we are a PFIC.
Taxation of Dividends Paid on Common Shares
We do not intend to pay dividends
in the foreseeable future. In the event that we do pay dividends, and subject to the discussion under the heading “Passive Foreign
Investment Companies” below and the discussion of “qualified dividend income” below, a U.S. Holder, other than certain
U.S. Holders that are U.S. corporations, will be required to include in gross income as ordinary income the amount of any distribution
paid on common shares (including the amount of any Swiss tax withheld on the date of the distribution), to the extent that such distribution
does not exceed our current and accumulated earnings and profits, as determined for U.S. federal income tax purposes. The amount of a
distribution which exceeds our earnings and profits will be treated first as a non-taxable return of capital, reducing the U.S. Holder’s
tax basis for the common shares to the extent thereof, and then capital gain. We do not expect to maintain calculations of our earnings
and profits under U.S. federal income tax principles and, therefore, U.S. Holders should expect that the entire amount of any distribution
generally will be reported as dividend income.
In general, preferential tax
rates for “qualified dividend income” and long-term capital gains are applicable for U.S. Holders that are individuals, estates
or trusts. For this purpose, “qualified dividend income” means, inter alia, dividends received from a “qualified foreign
corporation.” A “qualified foreign corporation” is a corporation that is entitled to the benefits of a comprehensive
tax treaty with the United States which includes an exchange of information program. The IRS has stated that the Switzerland/U.S. Tax
Treaty satisfies this requirement and we believe we are eligible for the benefits of that treaty.
In addition, our
dividends will be qualified dividend income if our common shares are readily tradable on Nasdaq or another established securities
market in the United States. Dividends will not qualify for the preferential rate if we are treated, in the year the dividend is
paid or in the prior year, as a PFIC, as described below under “Passive Foreign Investment Companies.” A U.S. Holder
will not be entitled to the preferential rate: (1) if the U.S. Holder has not held our common shares for at least 61 days of the
121-day period beginning on the date which is 60 days before the ex-dividend date, or (2) to the extent the U.S. Holder is under an
obligation to make related payments on substantially similar property. Any days during which the U.S. Holder has diminished its risk
of loss on our common shares are not counted towards meeting the 61-day holding period. Finally, U.S. Holders who elect to treat the
dividend income as “investment income” pursuant to Code section 163(d)(4) will not be eligible for the preferential rate
of taxation.
The amount of a distribution
with respect to our common shares will be measured by the amount of the fair market value of any property distributed, and for U.S. federal
income tax purposes, the amount of any Swiss taxes withheld therefrom. Cash distributions paid by us in CHF will be included in the income
of U.S. Holders at a U.S. dollar amount based upon the spot rate of exchange in effect on the date the dividend is includible in the income
of the U.S. Holder, and U.S. Holders will have a tax basis in such CHF for U.S. federal income tax purposes equal to such U.S. dollar
value. If the U.S. Holder subsequently converts the CHF into U.S. dollars or otherwise disposes of it, any subsequent gain or loss in
respect of such CHF arising from exchange rate fluctuations will be U.S. source ordinary exchange gain or loss.
Except as described below
in “Passive Foreign Investment Companies,” ownership of the Warrants has no tax impact on U.S. Holders since holders of the
Warrants do not receive distributions unless and until the Warrants are exercised and common shares are purchased. As described below,
if we are a PFIC, ownership of the Warrants may impact the holding period of PFIC shares and impact elections under the PFIC tax rules.
Taxation of the Disposition of Common Shares
and Warrants
Except as provided under the
PFIC rules described below under “Passive Foreign Investment Companies,” upon the sale, exchange or other disposition of our
common shares or the Warrants, a U.S. Holder will recognize capital gain or loss in an amount equal to the difference between such U.S.
Holder’s tax basis for the common shares and Warrants in U.S. dollars and the amount realized on the disposition in U.S. dollar
(or its U.S. dollar equivalent determined by reference to the spot rate of exchange on the date of disposition, if the amount realized
is denominated in a foreign currency). The gain or loss realized on the sale, exchange or other disposition of common shares and Warrants
will be long-term capital gain or loss if the U.S. Holder has a holding period of more than one year at the time of the disposition. Individuals
who recognize long-term capital gains may be taxed on such gains at reduced rates of tax. The deduction of capital losses is subject to
various limitations.
Passive Foreign Investment Companies
Special U.S. federal income
tax laws apply to U.S. taxpayers who own shares and warrants of a corporation that is a PFIC. We will be treated as a PFIC for U.S. federal
income tax purposes for any taxable year that either:
| ● | 75%
or more of our gross income (including our pro rata share of gross income for any company, in which we are considered to own 25% or more
of the shares by value), in a taxable year is passive; or |
| ● | At
least 50% of our assets, averaged over the year and generally determined based upon fair market value (including our pro rata share of
the assets of any company in which we are considered to own 25% or more of the shares by value) are held for the production of, or produce,
passive income. |
For this purpose, passive
income generally consists of dividends, interest, rents, royalties, annuities and income from certain commodities transactions and from
notional principal contracts. Cash is treated as generating passive income.
We do not believe that we
will be a PFIC for the current taxable year although we have not determined whether we will be a PFIC in the future. The tests for determining
PFIC status are applied annually, and it is difficult to make accurate projections of future income and assets which are relevant to this
determination. In addition, our PFIC status may depend in part on the market value of our common shares. Accordingly, there can be no
assurance that we currently are not or will not become a PFIC.
If we currently are or become
a PFIC, each U.S. Holder who has not elected to mark the shares to market (as discussed below), would, upon receipt of certain distributions
by us and upon disposition of our common shares and Warrants at a gain: (1) have such distribution or gain allocated ratably over the
U.S. Holder’s holding period for the common shares, as the case may be; (2) the amount allocated to the current taxable year and
any period prior to the first day of the first taxable year in which we were a PFIC would be taxed as ordinary income; and (3) the amount
allocated to each of the other taxable years would be subject to tax at the highest rate of tax in effect for the applicable class of
taxpayer for that year, and an interest charge for the deemed deferral benefit would be imposed with respect to the resulting tax attributable
to each such other taxable year. In addition, when shares and warrants of a PFIC are acquired by reason of death from a decedent that
was a U.S. Holder, the tax basis of such shares would not receive a step-up to fair market value as of the date of the decedent’s
death, but instead would be equal to the decedent’s basis if lower, unless all gain were recognized by the decedent. Indirect investments
in a PFIC may also be subject to these special U.S. federal income tax rules.
A U.S. Holder of our Warrants
is taxed in a manner similar to a U.S. holder of common shares if the holder realizes gain on the sale of the Warrants. If the holder
of the Warrants exercises the Warrants to purchase common shares, the holding period over which any income realized is allocated includes
the holding period of the Warrants. The U.S. Warrant holder is treated as a holder of PFIC stock taxable under the ordinary income allocation
and interest charge regime described above.
The PFIC rules described above
would not apply to a U.S. Holder who makes a QEF election for all taxable years that such U.S. Holder has held the common shares while
we are a PFIC, provided that we comply with specified reporting requirements. A U.S. Holder of our Warrants may not make a QEF election
regarding our Warrants. Instead, each U.S. Holder who has made such a QEF election is required for each taxable year that we are a PFIC
to include in income such U.S. Holder’s pro rata share of our ordinary earnings as ordinary income and such U.S. Holder’s
pro rata share of our net capital gains as long-term capital gain, regardless of whether we make any distributions of such earnings or
gain. In general, a QEF election is effective only if we make available certain required information. The QEF election is made on a shareholder-by-shareholder
basis and generally may be revoked only with the consent of the IRS. We do not intend to notify U.S. Holders if we believe we will be
treated as a PFIC for any tax year. In addition, we do not intend to furnish U.S. Holders annually with information needed in order to
complete IRS Form 8621 and to make and maintain a valid QEF election for any year in which we or any of our subsidiaries are a PFIC. Therefore,
the QEF election will not be available with respect to our common shares.
In addition, except with respect
to the Warrants (unless exercised), the PFIC rules described above would not apply if we were a PFIC and a U.S. Holder made a mark-to-market
election. A U.S. Holder of our common shares which are regularly traded on a qualifying exchange, including Nasdaq, can elect to mark
the common shares to market annually, recognizing as ordinary income or loss each year an amount equal to the difference as of the close
of the taxable year between the fair market value of the common shares and the U.S. Holder’s adjusted tax basis in the common shares.
Losses are allowed only to the extent of net mark-to-market gain previously included income by the U.S. Holder under the election for
prior taxable years.
U.S. Holders who hold our
common shares and Warrants during a period when we are a PFIC will be subject to the foregoing rules, even if we cease to be a PFIC. U.S.
Holders are strongly urged to consult their tax advisors about the PFIC rules.
Tax on Net Investment Income
U.S. Holders who are individuals,
estates or trusts will generally be required to pay a 3.8% Medicare tax on their net investment income (including dividends on and gains
from the sale or other disposition of our common shares), or in the case of estates and trusts on their net investment income that is
not distributed. In each case, the 3.8% Medicare tax applies only to the extent the U.S. Holder’s total adjusted income exceeds
applicable thresholds.
Tax Consequences for Non-U.S. Holders of Common
Shares and Warrants
Except as provided below,
an individual, corporation, estate or trust that is not a U.S. Holder referred to below as a non-U.S. Holder, generally will not be subject
to U.S. federal income or withholding tax on the payment of dividends on, and the proceeds from the disposition of, our common shares.
A non-U.S. Holder may be subject
to U.S. federal income tax on a dividend paid on our common shares (but not Warrants) or gain from the disposition of our common shares
and Warrants if: (1) such item is effectively connected with the conduct by the non-U.S. Holder of a trade or business in the United States
and, if required by an applicable income tax treaty is attributable to a permanent establishment or fixed place of business in the United
States; or (2) in the case of a disposition of our common shares and Warrants, the individual non-U.S. Holder is present in the United
States for 183 days or more in the taxable year of the disposition and other specified conditions are met.
In general, non-U.S. Holders
will not be subject to backup withholding with respect to the payment of dividends on our common shares if payment is made through a paying
agent, or office of a foreign broker outside the United States. However, if payment is made in the United States or by a U.S. related
person, non-U.S. Holders may be subject to backup withholding, unless the non-U.S. Holder provides an applicable IRS Form W-8 (or a substantially
similar form) certifying its foreign status, or otherwise establishes an exemption.
The amount of any backup withholding
from a payment to a non-U.S. Holder will be allowed as a credit against such holder’s U.S. federal income tax liability and may
entitle such holder to a refund, provided that the required information is timely furnished to the IRS.
Information Reporting and Withholding
A U.S. Holder may be subject
to backup withholding at a rate of 24% with respect to cash dividends and proceeds from a disposition of common shares. In general, backup
withholding will apply only if a U.S. Holder fails to comply with specified identification procedures. Backup withholding will not apply
with respect to payments made to designated exempt recipients, such as corporations and tax-exempt organizations. Backup withholding is
not an additional tax and may be claimed as a credit against the U.S. federal income tax liability of a U.S. Holder, provided that the
required information is timely furnished to the IRS.
A U.S. Holder with interests
in “specified foreign financial assets” (including, among other assets, our common shares, unless such common shares are held
on such U.S. Holder’s behalf through a financial institution) may be required to file an information report with the IRS if the
aggregate value of all such assets exceeds $50,000 on the last day of the taxable year or $75,000 at any time during the taxable year
(or such higher dollar amount as may be prescribed by applicable IRS guidance); and may be required to file a Report of Foreign Bank and
Financial Accounts if the aggregate value of the foreign financial accounts exceeds $10,000 at any time during the calendar year. You
should consult your own tax advisor as to the possible obligation to file such information report.
F. Dividends and Paying Agents
Not applicable.
G. Statement by Experts
Not applicable.
H. Documents on Display
We are subject to
certain information reporting requirements of the Exchange Act, applicable to foreign private issuers and under those requirements
will file reports with the SEC. The SEC maintains an Internet website that contains reports and other information regarding issuers
that file electronically with the SEC. Our filings with the SEC will also available to the public through the SEC’s website at
www.sec.gov.
As a foreign private issuer,
we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors
and principal shareholders will be exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the
Exchange Act. In addition, we are not required under the Exchange Act to file annual, quarterly and current reports and financial statements
with the SEC as frequently or as promptly as U.S. domestic companies whose securities are registered under the Exchange Act. However,
we will file with the SEC, within 120 days after the end of each fiscal year, or such applicable time as required by the SEC, an annual
report containing financial statements audited by an independent registered public accounting firm, and may submit to the SEC, on a Form
6-K, unaudited half-yearly financial information.
We maintain a corporate website
https://nlspharma.com/. Information contained on, or that can be accessed through, our website and the other websites referenced above
do not constitute a part of this annual report. We have included these website addresses in this annual report solely as inactive textual
references.
I. Subsidiary Information.
Our wholly owned subsidiaries
include NLS Pharmaceutics Inc., a Delaware corporation incorporated on April 27, 2021, and NLS Pharmaceutics (Israel) Ltd., an Israeli
company incorporated on November 3, 2024.
J. Annual Report to Security Holders.
Not applicable.
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES
ABOUT MARKET RISK.
We are exposed to market risks
in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position due to adverse
changes in financial market prices and rates. Our current investment policy is to invest available cash in bank deposits with banks that
have a credit rating of at least A-. Accordingly, a substantial majority of our cash and cash equivalents is held in deposits that bear
interest. Given the current low rates of interest we receive, we will not be adversely affected if such rates are reduced. Our market
risk exposure is primarily a result of foreign currency exchange rates, which is discussed in detail in the following paragraph.
Foreign Currency Exchange Risk
Our results of operations
and cash flows are subject to fluctuations due to changes in foreign currency exchange rates. As discussed above, the vast majority of
our liquid assets is held in USD, and a certain portion of our expenses are denominated in CHF or EUR. For instance, during the year ended
December 31, 2023, approximately 26% of our expenses were denominated in CHF, approximately 7% in EUR and 1% in GBP. Changes of 5% and
10% in the USD/CHF exchange rate would have increased/decreased our operating expenses by 1% and 2%, respectively. However, these historical
figures may not be indicative of future exposure, as we expect that the percentage of our CHF denominated expenses will materially decrease
in the near future, therefore reducing our exposure to exchange rate fluctuations.
We do not hedge our foreign
currency exchange risk. In the future, we may enter into formal currency hedging transactions to decrease the risk of financial exposure
from fluctuations in the exchange rates of our principal operating currencies. These measures, however, may not adequately protect us
from the material adverse effects of such fluctuations.
ITEM 12. DESCRIPTION OF SECURITIES OTHER THAN
EQUITY SECURITIES
A. Debt Securities.
Not applicable.
B. Warrants and rights.
Not applicable.
C. Other Securities.
Not applicable.
D. American Depositary Shares.
Not applicable.
PART II
ITEM 13. DEFAULTS, DIVIDEND ARREARAGES AND
DELINQUENCIES
Not applicable.
ITEM 14. MATERIAL MODIFICATIONS TO THE RIGHTS
OF SECURITY HOLDERS AND USE OF PROCEEDS
E. Use of Proceeds
Not applicable.
ITEM 15. CONTROLS AND PROCEDURES
(a) Disclosure Controls and Procedures
We maintain disclosure controls
and procedures designed to provide reasonable assurance that information required to be disclosed in reports filed under the Exchange
Act is recorded, processed, summarized and reported within the specified time periods and accumulated and communicated to our management,
including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Our management, with the participation
of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures
(as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of December 31, 2024. As of December 31, 2024, our
disclosure controls and procedures were not effective as a result of our material weaknesses in our internal control over financial reporting.
(b) Management’s Annual Report on Internal
Control over Financial Reporting
Our management is
responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial
reporting is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act as a process designed by, or under the supervision of,
a company’s principal executive officer and principal financial officer, or persons performing similar functions, and effected
by a company’s board of directors, management, and other personnel, to provide reasonable assurance regarding the reliability
of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted
accounting principles and includes those policies and procedures that:
| ● | pertain
to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of a company’s
assets; |
| ● | provide
reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally
accepted accounting principles, and that a company’s receipts and expenditures are being made only in accordance with authorizations
of the company’s management and directors; and |
| ● | provide
reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could
have a material effect on the financial statements. |
Because of its inherent limitations,
internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance
with the policies or procedures may deteriorate.
As described in Item 3.D.
“Risk Factors,” as of December 31, 2024, we have material weaknesses in our internal control over financial reporting relating
to a lack of sufficient number of trained professionals with an appropriate level of accounting knowledge to design and maintain controls
over the preparation of financial statements, and relating to a lack of maintaining appropriate segregation of duties.
Under the supervision of and
with the participation of our principal executive officer and principal financial officer, our management assessed the effectiveness of
our internal control over financial reporting as of December 31, 2024, based on the criteria set forth by the Committee of Sponsoring
Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013). Based on this assessment, management concluded
that our internal control over financial reporting was not effective as of December 31, 2024. We have taken steps to remediate the material
weaknesses and to strengthen our internal control over financial reporting. We conducted an analysis to identify the underlying reasons
for the material weaknesses. This involved assessing risk factors, understanding process gaps, and pinpointing areas that needed improvement.
As part of our remediation, we have transitioned the accounting processes to a dedicated team with U.S. GAAP and SEC reporting knowledge,
with the support of additional external advisors. While we believe that these efforts will improve our internal control over financial
reporting in accordance with U.S. GAAP and SEC reporting requirements, the implementation of these measures is ongoing and will require
validation and testing of the design and operating effectiveness of internal controls over a sustained period of financial reporting cycles.
This change strengthens our internal controls and ensures compliance with US GAAP.
(c) Attestation Report of the Registered Public
Accounting Firm
This annual report does not
include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting
due to an exemption for EGCs provided in the JOBS Act.
(d) Changes in Internal Control over Financial
Reporting
During the year ended December
31, 2024, we have taken steps towards remediating the material weaknesses and to strengthen our internal control over financial reporting,
by transitioning the accounting processes to a dedicated team with U.S. GAAP and SEC reporting knowledge, with the support of additional
external advisors. This measure was implemented to enhance the effectiveness and reliability of our financial reporting. We have appointed
a Chief Financial Officer with U.S. GAAP and SEC reporting knowledge to lead the internal preparation of half-yearly and yearly financial
statements, enabling us to identify and evaluate accounting treatment of any new activity and publish our half-yearly and yearly financial
statements. The implementation of these measures is ongoing and will require validation and testing of the design and operating effectiveness
of internal controls over a sustained period of financial reporting cycles. We cannot assure you that the measures we have taken to date,
and are continuing to implement, will be sufficient to establish and maintain effective internal control over financial reporting.
ITEM 16. [RESERVED.]
ITEM 16A. AUDIT COMMITTEE FINANCIAL EXPERT
Our board of directors has
determined that Mr. Gian-Marco Rinaldi, a member of our audit committee is an audit committee financial expert, as defined under the rules
under the Exchange Act, and is independent in accordance with applicable Exchange Act rules and Nasdaq rules.
ITEM 16B. CODE OF ETHICS
Code of Business Conduct and Ethics
Pursuant to Swiss law, we
adopted a code of business conduct and ethics which covers a broad range of matters including the handling of conflicts of interest, compliance
issues and other corporate policies such as insider trading and equal opportunity and non-discrimination standards. Our code of business
conduct and ethics is applicable to all our directors, senior management and employees. We have published our code of business conduct
and ethics on our website, www.nlspharma.com. If we make any amendment to the code of business conduct and ethics or grant any
waivers, including any implicit waiver, from a provision of the code, we will disclose the nature of such amendment or waiver on our website
to the extent required by the rules and regulations of the SEC including the instructions to Item 16.B. of Form 20-F.
ITEM 16C. PRINCIPAL ACCOUNTANT FEES AND SERVICES
On June 27, 2024, our shareholders
approved the appointment of Marcum LLP, an independent registered public accounting firm, to serve as our statutory auditor for the year
ended December 31, 2024, after our former auditor PricewaterhouseCoopers AG, informed us that they would not seek reelection as our independent
registered public accounting firm. The following table provides information regarding fees billed by Marcum LLP, an independent registered
public accounting firm, and PricewaterhouseCoopers AG, independent registered public accounting firm, or PwC, including audit services,
for the years ended December 31, 2024 and 2023.
| | |
Year Ended December 31, | |
| | |
2024 | | |
2023(3) | |
| Audit fees (1) | |
$ | 177,886 | | |
$ | 332,690 | |
| Audit-related fees (2) | |
| 121,663 | | |
| - | |
| Tax fees | |
| - | | |
| - | |
| All other fees | |
| | | |
| - | |
| Total | |
$ | 299,549 | | |
$ | 332,690 | |
| (1) |
Consists of fees billed for the audit of our annual financial statements, review of financial statements included in our services that are normally provided by the auditors in connection with statutory and regulatory filings or engagements. |
| |
|
| (2) |
This includes audit-related fees paid to PwC for the year ended
December 31, 2024. Audit-related fees relate to work regarding preparation of our Registration Statement on Form F-1, resale
Registration Statement on Form F-3 and our Merger-related Registration Statement on Form F-4 and amendments thereto.
|
| |
|
| (3) |
PwC only. |
Pre-Approval of Auditors’ Compensation
Our audit committee has a
pre-approval policy for the engagement of our independent registered public accounting firm to perform certain audit and non-audit services.
Pursuant to this policy, which is designed to assure that such engagements do not impair the independence of our auditors, the audit committee
pre-approves annually a catalog of specific audit and non-audit services in the categories of audit services, audit-related services and
tax services that may be performed by our independent registered public accounting firm. If a type of service, that is to be provided
by our auditors, has not received such general pre-approval, it will require specific pre-approval by our audit committee. The policy
prohibits retention of the independent registered public accounting firm to perform the prohibited non-audit functions defined in applicable
SEC rules.
ITEM 16D. EXEMPTIONS FROM THE LISTING STANDARDS
FOR AUDIT COMMITTEES
Not applicable.
ITEM 16E. PURCHASES OF EQUITY SECURITIES BY
THE ISSUER AND AFFILIATED PURCHASERS
Not applicable.
ITEM 16F. CHANGE IN REGISTRANT’S CERTIFYING
ACCOUNTANT
Not applicable.
ITEM 16G. CORPORATE GOVERNANCE
Differences between Swiss Laws and Nasdaq
Requirements
The Sarbanes-Oxley Act, as
well as related rules subsequently implemented by the SEC, require foreign private issuers, such as us, to comply with various corporate
governance practices. In addition, we are required to comply with Nasdaq rules. Under those rules, we may elect to follow certain corporate
governance practices permitted under the Swiss law in lieu of compliance with corresponding corporate governance requirements otherwise
imposed by Nasdaq rules for U.S. domestic registrants.
In accordance with Swiss law
and practice and subject to the exemption set forth in Rule 5615 of the Nasdaq rules, as a foreign private issuer, we have elected to
rely on home country governance requirements and certain exemptions thereunder rather than the Nasdaq rules, with respect to the following
requirements:
| |
● |
Composition of the board of directors. Swiss law does not require that a majority of our board of directors consist of independent directors. Our board of directors therefore may include fewer independent directors than would be required if we were subject to Nasdaq Listing Rule 5605(b)(1). In addition, we will not be subject to Nasdaq Listing Rule 5605(b)(2), which requires that independent directors must regularly have scheduled meetings at which only independent directors are present. |
| |
● |
Quorum. In accordance with Swiss law and
generally accepted business practices, our articles of association do not provide quorum requirements generally applicable to general
meetings of shareholders. Our practice thus varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide
in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock.
According to Swiss law, for the approval of the
management report and the annual financial statements, the Company’s auditor must be present at such a general meeting of shareholders,
unless the general meeting of shareholders waives such attendance by unanimous decision. |
| |
● |
Proxy Solicitations. There are currently three types of institutional proxies under Swiss Law: (i) the independent proxy, (ii) the board proxy and (iii) the custodian proxy. According to Swiss Law, the solicitation of proxies by our board of directors or a custodian thereof is not permitted and only independent proxies are permitted for listed companies. The independent proxy has to be elected by the general meeting of shareholders, must only vote pursuant to the specific instructions of the shareholders and must abstain from voting in the absence of specific instructions. However, Swiss law does not have a regulatory regime for the solicitation of proxies and company solicitation of proxies is prohibited for public companies in Switzerland; thus our practice will vary from the requirement of Nasdaq Listing Rule 5620(b), which sets forth certain requirements regarding the solicitation of proxies. |
| |
● |
Share Issuances. Pursuant to Swiss law, prior to our initial public offering, we opted out of shareholder approval requirements by way of including authorized and conditional share capital for the issuance of securities in connection with certain events such as the acquisition of stock or assets of another company, the establishment of or amendments to equity-based compensation plans for employees, a change of control of the Company and certain private placements. Due to the revision of Swiss corporate law, effective January 1, 2023, or the Revision, we implemented the capital band (Kapitalband) in lieu of the existing authorized capital. To this extent, our practice varies from the requirements of Nasdaq Listing Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events. |
| |
● |
Compensation, Nomination and Governance Committee. Swiss law requires that we adopt a compensation committee, so in accordance with Nasdaq Listing Rule 5615(a)(3), we will follow home country requirements with respect to the compensation committee. Under home country rules, the general meeting of shareholders has the authority to set up the basic principles of the compensation committee’s responsibilities in a company’s articles of association. In addition, the shareholders may decide to include in the articles of association a list of concrete responsibilities of the compensation committee. Unless the articles of association list concrete responsibilities of the compensation committee, the determination of the concrete responsibilities falls within the competence of the board of directors. Furthermore, the general meeting of shareholders must vote on the compensation report and the total of the remunerations of the board of directors, senior management and advisory committee. According to the doctrine, the strategic planning of the remuneration system and policy as well as the determination of the remuneration procedure is the inalienable task of the board of directors, unless a list of concrete responsibilities is included in the articles of association of the Company. Apart from that, the board of directors has the authority to determine whether the compensation committee shall have the decision-making power with regard to the individual salaries or whether it shall have only a request right to the board of directors. |
Furthermore, pursuant to Swiss law,
both the number of committee members (the minimum and maximum number of compensation committee members) and the identity of the compensation
committee members is determined by our shareholders. Our home country rules also do not require us to authorize a committee of our independent
directors or alternatively hold a vote consisting of solely our independent directors in order to determine which persons shall be nominated
for election by our shareholders. Our practice will therefore vary from the requirements of Nasdaq Listing Rule 5605(d), which sets forth
certain requirements as to the responsibilities, composition and independence of compensation committees, and from the independent director
oversight of director nominations requirements of Nasdaq Listing Rule 5605(e). Furthermore, we are subject to the “Say on Pay”
Rule in accordance with Swiss law. This means that the compensation of our board of directors and senior management must be presented
by the board of directors to our shareholders and our shareholders must vote on the proposed compensation. Furthermore, pursuant to Swiss
law, severance, certain compensations in connection with non-competes, advances, transaction premiums and similar payments to senior management
and director are prohibited.
| |
● |
Code of Business Conduct and Ethics. Pursuant to Swiss law, we adopted a code of business conduct and ethics applicable to all of our directors, senior management and employees, which may not comply with the requirements of Nasdaq Listing Rule 5610. |
ITEM 16H. MINE SAFETY DISCLOSURE
Not applicable.
ITEM 16I. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT
INSPECTIONS
Not applicable.
ITEM 16J. INSIDER TRADING POLICIES
We have adopted an insider
trading policy, or the Policy, governing the purchase, sale and other transactions in our securities that applies to our directors, senior
management, employees, and other covered persons, including immediate family members and entities controlled by any of the foregoing persons.
The Policy prohibits, among
other things, insider trading and certain speculative transactions in our securities (including short sales, buying put and selling call
options and other hedging or derivative transactions in our securities) and establishes a regular blackout period schedule during which
directors, senior management, employees, and other covered persons may not trade in the Company’s securities, as well as certain
pre-clearance procedures that directors and senior management must observe prior to effecting any transaction in our securities.
The Company believes that
the Policy is reasonably designed to promote compliance with applicable insider trading laws, rules and regulations, and listing standards
applicable to the Company. A copy of the Policy is filed as Exhibit 11(b) to this Form 20-F.
ITEM 16K. CYBERSECURITY
Our board of directors recognizes
the critical importance of maintaining the trust and confidence of our customers, clients, business partners and employees. The audit
committee is involved in oversight of our IT general controls and cybersecurity. Our IT general controls cybersecurity policies, standards,
processes and practices are fully integrated into our risk management program. In general, we seek to address IT general controls cybersecurity
risks through a comprehensive, cross-functional approach that is focused on preserving the confidentiality, security and availability
of the information that we collect and store by identifying, preventing and mitigating cybersecurity threats and effectively responding
to cybersecurity incidents when they occur. In that regard, we have a comprehensive IT policy that outlines our security risk
measures. In addition, we have partnered with a professional IT service provider who specializes in security, endpoint protection,
and monitoring. We also operate the “Microsoft Office 365 E3” plan.
Risk Management and Strategy
As part of the critical elements
of our overall risk management approach, our cybersecurity program is focused on the following key areas:
| |
● |
Governance: As discussed in more detail under the heading “Governance,” the audit committee oversees our cybersecurity risk management and interacts with the Company’s Sarbanes-Oxley Act, or SOX, advisor and the Company’s Information Security advisor and other relevant members of management. |
| |
● |
Collaborative Approach: The Company has implemented a comprehensive, cross-functional approach to identifying, preventing and mitigating cybersecurity threats and incidents, while also implementing controls and procedures that provide for the prompt escalation of certain cybersecurity incidents so that decisions regarding the public disclosure and reporting of such incidents can be made by management in a timely manner. |
| |
● |
Technical Safeguards: The Company deploys technical safeguards that are designed to protect the Company’s information systems from cybersecurity threats, including firewalls, intrusion prevention and detection systems, anti-malware functionality and access controls, which are evaluated and improved through vulnerability assessments and cybersecurity threat intelligence. |
| |
● |
Recovery Planning: The Company has established and maintains comprehensive recovery plans that fully address the Company’s response to a cybersecurity incident, and such plans are tested and evaluated on a regular basis. |
| |
● |
Education and Awareness: The Company provides regular, mandatory training for personnel regarding cybersecurity threats as a means to equip the Company’s personnel with effective tools to address cybersecurity threats, and to communicate the Company’s evolving information security policies, standards, processes and practices. |
We engage in the periodic
assessment and testing of our policies, standards, processes and practices that are designed to address cybersecurity threats and incidents.
These efforts include a wide range of activities, including audits, assessments, tabletop exercises, threat modeling, vulnerability testing,
and other exercises focused on evaluating the effectiveness of our cybersecurity measures and planning. We are engaged with a subcontractor
that monitors the IT system and cybersecurity, as part of their routine job functions. We regularly engage third parties to perform assessments
on our cybersecurity measures, including information security maturity assessments, audits and independent reviews of our information
security control environment and operating effectiveness. The results of such assessments, audits and reviews are reported to the Board,
and we adjust our cybersecurity policies, standards, processes and practices as necessary based on the information provided by these assessments,
audits and reviews
Governance
The audit committee interacts
with the Company’s SOX advisor and the Company’s Information Security advisor and other relevant members of management. The
Company’s Information Security advisor is responsible for assessing and managing cyber risk management, informs senior management
regarding the prevention, detection, mitigation, and remediation of cybersecurity incidents and supervises such efforts. The Company’s
Information Security advisor has experience selecting, deploying, and operating cybersecurity technologies, initiatives, and processes,
and relies on threat intelligence as well as other information obtained from governmental, public or private sources. The audit committee
receives updates on IT general controls, and a set of policies and procedures that govern how the Company’s IT systems operate and
ensure the confidentiality, integrity, and availability of data as well as on cybersecurity risks, including prevention of data theft,
unauthorized access, operational disruption, and data breaches. The audit committee also receives prompt and timely information regarding
any cybersecurity incident that meets established reporting thresholds, as well as ongoing updates regarding any such incident until it
has been addressed. On an annual basis, the audit committee discusses our approach to IT general controls and cybersecurity risk management
with the Company’s SOX advisor.
As of the date of this annual
report, we do not believe that we have experienced any cybersecurity attack. Cybersecurity threats and cybersecurity incidents, if they
occur, can materially affect or are reasonably likely to affect the Company, including its business strategy, results of operations or
financial condition.
PART III
ITEM 17. FINANCIAL STATEMENTS
We have elected to provide
financial statements and related information pursuant to Item 18.
ITEM 18. FINANCIAL STATEMENTS
The consolidated financial
statements and the related notes required by this Item are included in this annual report beginning on page F-1.
ITEM 19. EXHIBITS
The following documents are filed as part of the
annual report.
| Exhibit Number |
|
Exhibit Description |
| 1.1 |
|
Amended and Restated Articles of Association of NLS Pharmaceutics Ltd. (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed on November 15, 2024). |
| 2.1* |
|
Description of Securities |
| 4.1 |
|
Share Option Plan Regulation 2021 (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed on December 22, 2021). |
| 4.2 |
|
At-the-Market Sales Agreement, or the Sales Agreement, with Virtu Americas LLC, dated March 3, 2022 (filed as Exhibit 1.1 to Form 6-K (File No. 001-39957) filed on March 4, 2022). |
| 4.3 |
|
Form of Common Warrant (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed December 8, 2022). |
| 4.4 |
|
Form of Pre-Funded Warrant (filed as Exhibit 99.2 to Form 6-K (File No. 001-39957) filed December 8, 2022). |
| 4.5 |
|
Form of Common Warrant (filed as Exhibit 4.1 to Form 6-K File No. 001-39957) filed April 14, 2022). |
| 4.6 |
|
Form of Pre-Funded Warrant (filed as Exhibit 4.2 to Form 6-K (File No. 001-39957) filed April 14, 2022). |
| 4.7 |
|
Form of Short Term Note Agreement (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed August 23, 2022). |
| 4.8 |
|
Form of Common Warrant (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed October 3, 2022). |
| 4.9 |
|
Securities Purchase Agreement, by and between NLS Pharmaceutics Ltd. and Armistice Capital Master Fund Ltd. and Lind Global Fund II LP, dated March 20, 2024 (filed as Exhibit 10.1 to Form 6-K (File No. 001-39957) filed March 21, 2024). |
| 4.10 |
|
Form of Common Warrant (filed as Exhibit 4.1 to Form 6-K (File No. 001-39957) filed March 21, 2024). |
| 4.11 |
|
Form of Placement Agent Warrant (filed as Exhibit 4.2 to Form 6-K (File No. 001-39957) filed March 21, 2024). |
| 4.12 |
|
Securities Purchase Agreement, by and between NLS Pharmaceutics Ltd. and Armistice Capital Master Fund Ltd. and Intracoastal Capital LLC, dated June 28, 2024 (filed as Exhibit 10.1 to Form 6-K (File No. 001-39957) filed July 1, 2024). |
| 4.13 |
|
Form of Common Warrant (filed as Exhibit 4.1 to Form 6-K (File No. 001-39957) filed July 1, 2024). |
| 4.14 |
|
Form of Series A Placement Agent Warrant (filed as Exhibit 4.2 to Form 6-K (File No. 001-39957) filed July 1, 2024). |
| 4.15 |
|
Form of Series B Placement Agent Warrant (filed as Exhibit 4.3 to Form 6-K (File No. 001-39957) filed July 1, 2024). |
| 4.16 |
|
Form of Warrant Amendment Agreement, by and between NLS Pharmaceutics Ltd. and Armistice Capital Master Fund Ltd., dated September 16, 2024 (filed as Exhibit 10.1 to Form 6-K (File No. 001-39957) filed September 16, 2024). |
| 4.17 |
|
Form of Pre-Funded Warrant (filed as Exhibit 10.2 to Form 6-K (File No. 001-39957) filed September 16, 2024). |
| 4.18 |
|
Form of Equity Securities Purchase Agreement, dated October 9, 2024, by and among NLS Pharmaceutics and certain purchasers thereto (filed as Exhibit 99.2 to Form 6-K (File No. 001-39957) filed October 11, 2024). |
| 4.19 |
|
Form of Debt Securities Purchase Agreement, dated October 9, 2024, by and among NLS Pharmaceutics and certain purchasers thereto (filed as Exhibit 99.3 to Form 6-K (File No. 001-39957) filed October 11, 2024). |
| 4.20 |
|
Form of Common Warrant (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed October 11, 2024). |
| 4.21 |
|
Form of Pre-Funded Warrant (filed as Exhibit 99.5 to Form 6-K (File No. 001-39957) filed October 11, 2024). |
| 4.22 |
|
Form of Warrant Amendment Agreement (filed as Exhibit 99.4 to Form 6-K (File No. 001-39957) filed October 11, 2024). |
| 4.23 |
|
Form of Registration Rights Agreement (equity) (filed as Exhibit 99.6 to Form 6-K (File No. 001-39957) filed October 11, 2024). |
| 4.24 |
|
Form of Registration Rights Agreement (debt) (filed as Exhibit 99.7 to Form 6-K (File No. 001-39957) filed October 11, 2024). |
| 4.25 |
|
Form of Exchange Agreement (filed as Exhibit 99.8 to Form 6-K (File No. 001-39957) filed October 11, 2024). |
| 4.26 |
|
Agreement and Plan of Merger, dated as of November 4, 2024, by and among NLS Pharmaceutics Ltd., NLS Pharmaceutics (Israel) Ltd. and Kadimastem Ltd. (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed November 5, 2024). |
| 4.27 |
|
First Amendment to Agreement and Plan of Merger, by and among NLS Pharmaceutics Ltd., NLS Pharmaceutics (Israel) Ltd., and Kadimastem Ltd., dated January 30, 2025 (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed January 30, 2025). |
| 4.28* |
|
Second Amendment to Agreement and Plan of Merger, by and among NLS Pharmaceutics Ltd., NLS Pharmaceutics (Israel) Ltd., and Kadimastem Ltd., dated February 17, 2025. |
| 4.29 |
|
Third Amendment to Agreement and Plan of Merger, by and among NLS Pharmaceutics Ltd., NLS Pharmaceutics (Israel) Ltd., and Kadimastem Ltd., dated May 5, 2025 (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed May 5, 2025). |
| 4.30 |
|
Form of Contingent Value Rights Agreement,by and between NLS Pharmaceutics Ltd. and VStock Transfer, LLC, dated November 4, 2024 (filed as Exhibit 99.2 to Form 6-K (File No. 001-39957) filed November 5, 2024). |
| 4.31 |
|
Form of Support Agreement, by and between NLS Pharmaceutics Ltd. and certain shareholders of Kadimastem Ltd., dated November 4, 2024 (filed as Exhibit 99.3 to Form 6-K (File No. 001-39957) filed November 5, 2024). |
| 4.32 |
|
Securities Purchase Agreement, by and between NLS Pharmaceutics Ltd. and Alpha Capital Anstalt, dated December 4, 2024 (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed December 5, 2024). |
| 4.33 |
|
Securities Purchase Agreement, by and between NLS Pharmaceutics Ltd. and certain accredited investors, dated March 27, 2025 (filed as Exhibit 99.1 to Form 6-K (File No. 001-39957) filed March 28, 2025). |
| 4.34 |
|
Common Shares Purchase Agreement, by and between NLS Pharmaceutics Ltd. and an institutional investor, dated March 31, 2025 (filed as Exhibit 99.2 to Form 6-K (File No. 001-39957) filed April 1, 2025). |
| 4.35 |
|
Form of Pre-Funded Warrant (filed as Exhibit 99.4 to Form 6-K (File No. 001-39957) filed March 31, 2025). |
| 4.36 |
|
Registration Rights Agreement, by and between NLS Pharmaceutics Ltd. and the investors party thereto, dated March 27, 2025 (filed as Exhibit 99.2 to Form 6-K (File No. 001-39957) filed March 31, 2025). |
| 4.37 |
|
License Agreement by and between NLS Pharmaceutics Ltd. and Novartis Pharma AG, dated March 10, 2021 (filed as Exhibit 4.6 to Annual Report on Form 20-F filed on May 14, 2021). |
| 4.38 |
|
Amendment No. 1 to Pre-Funded Warrant dated April 26, 2023 between NLS Pharmaceutics Ltd. and the pre-funded warrant holders named therein (filed as Exhibit 4.17 to Annual Report on Form 20-F filed May 15, 2024). |
| 4.39 |
|
License Agreement by and between NLS Pharmaceutics Ltd. and Aexon Labs Inc., dated March 19, 2024 (filed as Exhibit 4.18 to Annual Report on Form 20-F (File No. 001-39957) filed May 15, 2024). |
| 4.40 |
|
Addendum to Loan Agreement dated November 15, 2023 (filed as Exhibit 99.2 to Form 6-K filed May 14, 2024). |
| 4.41 |
|
Second Addendum to Loan Agreement dated September 28, 2023 (filed as Exhibit 99.1 to Form 6-K filed May 14, 2024). |
| 8.1* |
|
Listing of Subsidiaries (filed as Exhibit 8.1 to Annual Report on Form 20-F filed on May 14, 2021). |
| 11(b)* |
|
Insider Trading Policy |
| 12.1* |
|
Certification of the Chief
Executive Officer pursuant to rule 13a-14(a) of the Securities Exchange Act of 1934 (filed herewith). |
| 12.2* |
|
Certification of the Chief
Financial Officer pursuant to rule 13a-14(a) of the Securities Exchange Act of 1934 (filed herewith). |
| 13.1** |
|
Certification of the Chief
Executive Officer pursuant to 18 U.S.C. 1350, furnished herewith. |
| 13.2** |
|
Certification of the Chief
Financial Officer pursuant to 18 U.S.C. 1350, furnished herewith. |
| 15.1* |
|
Consent of PricewaterhouseCoopers AG. |
| 15.2* |
|
Consent of Marcum LLP. |
| 97.1 |
|
Clawback Policy (filed as Exhibit 97.1 to Annual Report on Form 20-F filed May 15, 2024). |
| 101* |
|
The following financial information from the Registrant’s Annual Report on Form 20-F for the year ended December 31, 2023, formatted in XBRL (eXtensible Business Reporting Language): (i) Balance Sheets; (ii) Statements of Operations and Comprehensive Loss; (iii) Statements of Changes in Shareholders’ Deficit; (iv) Statements of Cash Flows; and (v) Notes to the financial statements, tagged as blocks of text and in detail. |
| 104* |
|
Cover Page Interactive Data File (embedded within the Inline XBRL document). |
SIGNATURES
The registrant hereby certifies
that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this
annual report on Form 20-F filed on its behalf.
| |
NLS Pharmaceutics Ltd. |
| |
|
|
| Date May 16, 2025 |
By: |
/s/ Alexander Zwyer |
| |
|
Alexander Zwyer |
| |
|
Chief Executive Officer |
| |
|
|
| |
By: |
/s/ Nicole Fernandez-McGovern |
| |
|
Nicole Fernandez-McGovern |
| |
|
Chief Financial Officer |
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
CONSOLIDATED FINANCIAL STATEMENTS
YEARS ENDED DECEMBER 31, 2024, 2023 AND 2022
NLS PHARMACEUTICS LTD.
CONSOLIDATED FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting
Firm
To the Shareholders and Board of Directors of
NLS Pharmaceutics Ltd.
Opinion on the Financial Statements
We have audited the accompanying consolidated
balance sheet of NLS Pharmaceutics Ltd. (the “Company”) as of December 31, 2024, the related consolidated statements of operations
and comprehensive loss, stockholders’ equity and cash flows for the year then ended, and the related notes (collectively referred
to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the
financial position of the Company as of December 31, 2024, and the results of its operations and its cash flows for the year ended, in
conformity with accounting principles generally accepted in the United States of America.
Substantial Doubt about the Company’s
Ability to Continue as a Going Concern
The accompanying financial statements have been
prepared assuming that the Company will continue as a going concern. As more fully described in Note 1, the Company has incurred significant
losses and needs to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt
about the Company's ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1.
The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility
of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audit. We
are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are
required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and
regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the
standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial
statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged
to perform, an audit of its internal control over financial reporting. As part of our audit[we are required to obtain an understanding
of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal
control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess
the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond
to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements.
Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating
the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/ Marcum llp
Marcum llp
We have served as the Company’s auditor since 2024.
New York
May 16, 2025
Report of Independent Registered Public Accounting
Firm
To the Shareholders and Board of Directors of
NLS Pharmaceutics Ltd.
Opinion on the Financial Statements
We have audited the consolidated balance
sheet of NLS Pharmaceutics Ltd. (the “Company”) as of December 31, 2023, and the related consolidated statements of
operations and comprehensive loss, of changes in shareholders’ equity (deficit) and of cash flows for each of the two years in
the period ended December 31, 2023, including the related notes (collectively referred to as the “consolidated financial
statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial
position of the Company as of December 31, 2023, and the results of its operations and its cash flows for each of the two
years in the period ended December 31, 2023 in conformity with accounting principles generally accepted in the United States of
America.
Substantial Doubt about the Company’s
Ability to Continue as a Going Concern
The accompanying consolidated financial
statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated
financial statements, the Company has generated operating losses and expects to continue to generate operating losses and negative
operating cash flows and its existing cash and cash equivalents and access to existing financing arrangements will not be sufficient
to fund operations, that raise substantial doubt about its ability to continue as a going concern. Management’s plans in
regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that
might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are
the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s
consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting
Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S.
federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated
financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain
reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to
assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing
procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and
disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and
significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements.
We believe that our audits provide a reasonable basis for our opinion.
/s/ PricewaterhouseCoopers AG
Zurich, Switzerland
May 15, 2024, except for the effects of the reverse share split discussed in Note 2 to the consolidated financial statements, as to which
the date is May 16, 2025
We served as the Company’s auditor from 2021 to 2024.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
| | |
December 31, | |
| | |
2024 | | |
2023 | |
| ASSETS | |
| | |
| |
| Current assets: | |
| | |
| |
| Cash and cash equivalents | |
$ | 1,665,395 | | |
$ | 897,680 | |
| Prepaid expenses and other current assets (Note 3) | |
| 560,157 | | |
| 925,382 | |
| Total current assets | |
| 2,225,552 | | |
| 1,823,062 | |
| | |
| | | |
| | |
| Property and equipment, net | |
| 7,290 | | |
| 6,694 | |
| Other assets | |
| 283 | | |
| 16,885 | |
| Total assets | |
$ | 2,233,125 | | |
$ | 1,846,641 | |
| | |
| | | |
| | |
| LIABILITIES AND SHAREHOLDERS’ EQUITY (DEFICIT) | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Accounts payable, including related party of $15,000 and $265,864, as of December 31, 2024 and 2023, respectively | |
$ | 515,486 | | |
$ | 4,633,534 | |
| Related party short-term loans of $0 and $1,633,746, as of December 31, 2024, and 2023, respectively | |
| - | | |
| 1,633,746 | |
| Other accrued liabilities | |
| 311,278 | | |
| 1,652,270 | |
| Total current liabilities | |
| 826,764 | | |
| 7,919,550 | |
| | |
| | | |
| | |
| Deferred revenues | |
| - | | |
| 2,499,969 | |
| Accrued pension liability | |
| - | | |
| 260,685 | |
| Total liabilities | |
| 826,764 | | |
| 10,680,204 | |
| | |
| | | |
| | |
| Commitments and contingencies (Note 9) | |
| | | |
| | |
| | |
| | | |
| | |
| Shareholders’ equity (deficit): | |
| | | |
| | |
| Preferred shares, CHF 0.80 ($0.80) par value, 206,452 registered shares issued and outstanding at December 31, 2024 | |
| 2,740,958 | | |
| - | |
| Common shares, CHF 0.80 ($0.80) par value, 3,159,535 and 810,722 registered shares issued and outstanding at December 31, 2024 and 2023, respectively | |
| 166,353 | | |
| 668,555 | |
| Additional paid-in capital | |
| 72,820,671 | | |
| 61,029,437 | |
| Accumulated deficit | |
| (74,430,474 | ) | |
| (70,373,484 | ) |
| Accumulated other comprehensive loss | |
| 108,853 | | |
| (158,071 | ) |
| Total shareholders’ equity (deficit) | |
| 1,406,361 | | |
| (8,833,563 | ) |
| Total liabilities and shareholders’ equity (deficit) | |
$ | 2,233,125 | | |
$ | 1,846,641 | |
The accompanying notes are an integral part of
these consolidated financial statements.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE
LOSS
| | |
For the Years Ended December 31, | |
| | |
2024 | | |
2023 | | |
2022 | |
| OPERATIONS: | |
| | |
| | |
| |
| Operating expenses: | |
| | |
| | |
| |
| Research and development | |
$ | 422,051 | | |
$ | 5,908,288 | | |
$ | 8,976,643 | |
| General and administrative | |
| 3,214,224 | | |
| 5,898,775 | | |
| 6,505,721 | |
| Merger transaction costs | |
| 743,838 | | |
| - | | |
| - | |
| Total operating expenses | |
| 4,380,113 | | |
| 11,807,063 | | |
| 15,482,364 | |
| | |
| | | |
| | | |
| | |
| Operating loss | |
| (4,380,113 | ) | |
| (11,807,063 | ) | |
| (15,482,364 | ) |
| | |
| | | |
| | | |
| | |
| Other income (expense): | |
| | | |
| | | |
| | |
| Other income due to the termination of EF License Agreement | |
| 2,499,969 | | |
| - | | |
| - | |
| Other income (expense), net | |
| 49,351 | | |
| (219,812 | ) | |
| 10,045 | |
| Interest expense | |
| (45,054 | ) | |
| (119,920 | ) | |
| (95,211 | ) |
| Interest on related party loans | |
| (104,963 | ) | |
| (25,233 | ) | |
| (5,655 | ) |
| Loss on extinguishment of convertible notes | |
| - | | |
| - | | |
| (922,495 | ) |
| Total other income (expense) | |
| 2,399,303 | | |
| (364,965 | ) | |
| (1,013,316 | ) |
| | |
| | | |
| | | |
| | |
| Net loss | |
| (1,980,810 | ) | |
| (12,172,029 | ) | |
| (16,495,680 | ) |
| Deemed dividend | |
| (2,076,180 | ) | |
| - | | |
| - | |
| Net loss attributable to common stockholders | |
| (4,056,990 | ) | |
| (12,172,029 | ) | |
| (16,495,680 | ) |
| | |
| | | |
| | | |
| | |
| Basic and diluted net loss per common share (i) | |
$ | (2.63 | ) | |
$ | (12.75 | ) | |
$ | (33.52 | ) |
| | |
| | | |
| | | |
| | |
| Weighted average common shares used in computing basic and diluted net loss per common share (i) | |
| 1,543,283 | | |
| 954,401 | | |
| 492,066 | |
| | |
| | | |
| | | |
| | |
| COMPREHENSIVE LOSS: | |
| | | |
| | | |
| | |
| Net loss | |
$ | (1,980,810 | ) | |
$ | (12,172,029 | ) | |
$ | (16,495,680 | ) |
| Defined pension plan adjustments | |
| 266,924 | | |
| (107,280 | ) | |
| 100,948 | |
| Total comprehensive loss | |
$ | (1,713,887 | ) | |
$ | (12,279,309 | ) | |
$ | (16,394,732 | ) |
| | |
| | | |
| | | |
| | |
| Basic and diluted net loss per common share | |
$ | (2.63 | ) | |
$ | (12.75 | ) | |
$ | (33.52 | ) |
| | |
| | | |
| | | |
| | |
| Weighted average common shares used in computing basic and diluted net loss per common share | |
| 1,543,283 | | |
| 954,401 | | |
| 492,066 | |
| (i) | Adjusted for the effect of a 1-to-40 reverse share split that
was effective September 27, 2024 (See note 2). |
The accompanying notes are an integral part of
these consolidated financial statements.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY (DEFICIT)
FOR THE YEARS ENDED DECEMBER 31, 2024, 2023 AND
2022
| | |
Preferred Shares | | |
Common Shares | | |
Additional Paid in | | |
(Accumulated | | |
Accumulated Other Comprehensive | | |
| |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Capital | | |
Deficit) | | |
Loss | | |
Total | |
| BALANCE, January 1, 2022 | |
| - | | |
$ | - | | |
| 405,585 | | |
$ | 314,948 | | |
$ | 42,084,954 | | |
$ | (41,705,775 | ) | |
$ | (151,739 | ) | |
$ | 542,388 | |
| Issuance of common shares direct offering, net | |
| | | |
| - | | |
| 75,384 | | |
| 60,308 | | |
| 1,851,205 | | |
| - | | |
| - | | |
| 1,911,513 | |
| Issuance of warrants, net | |
| - | | |
| - | | |
| - | | |
| - | | |
| 2,047,208 | | |
| - | | |
| - | | |
| 2,047,208 | |
| Issuance of pre-funded warrants, net | |
| - | | |
| - | | |
| - | | |
| - | | |
| 5,348,882 | | |
| - | | |
| - | | |
| 5,348,882 | |
| Issuance of common shares in At-The-Market (ATM) financing | |
| - | | |
| - | | |
| - | | |
| 440 | | |
| 30,553 | | |
| - | | |
| - | | |
| 30,993 | |
| Exercise of pre-funded warrants | |
| - | | |
| - | | |
| - | | |
| 23,692 | | |
| - | | |
| - | | |
| - | | |
| 23,692 | |
| Issuance of common shares in private placement offerings, net | |
| - | | |
| - | | |
| 266,842 | | |
| 218,838 | | |
| 7,056,711 | | |
| - | | |
| - | | |
| 7,275,549 | |
| Conversion of convertible notes payable | |
| - | | |
| - | | |
| 62,911 | | |
| 50,329 | | |
| 2,422,287 | | |
| - | | |
| - | | |
| 2,472,616 | |
| Stock-based compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| 22,730 | | |
| - | | |
| - | | |
| 22,730 | |
| Defined pension plan adjustments | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 100,948 | | |
| 100,948 | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (16,495,680 | ) | |
| | | |
| (16,495,680 | ) |
| BALANCE, DECEMBER 31, 2022 | |
| - | | |
| - | | |
| 810,722 | | |
| 668,555 | | |
| 60,864,530 | | |
| (58,201,455 | ) | |
| (50,791 | ) | |
$ | 3,280,839 | |
| Stock based compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| 164,907 | | |
| - | | |
| - | | |
| 164,907 | |
| Defined pension plan adjustments | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (107,280 | ) | |
| (107,280 | ) |
| Net loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (12,172,029 | ) | |
| - | | |
| (12,172,029 | ) |
| BALANCE, DECEMBER 31, 2023 | |
| - | | |
| - | | |
| 810,722 | | |
| 668,555 | | |
| 61,029,437 | | |
| (70,373,484 | ) | |
| (158,071 | ) | |
$ | (8,833,563 | ) |
| Issuance of common shares in private placement offerings, net | |
| - | | |
| - | | |
| 1,063,396 | | |
| 922,391 | | |
| 4,330,334 | | |
| - | | |
| - | | |
| 5,252,725 | |
| Issuance of preferred shares for assumption of debt | |
| 806,452 | | |
| 708,969 | | |
| - | | |
| - | | |
| 3,291,031 | | |
| - | | |
| - | | |
| 4,000,000 | |
| Conversion of preferred shares into common shares | |
| (600,000 | ) | |
| (542,616 | ) | |
| 600,000 | | |
| 542,616 | | |
| - | | |
| - | | |
| - | | |
| - | |
| Issuance of common shares upon debt conversion | |
| - | | |
| - | | |
| 493,986 | | |
| 434,273 | | |
| 2,354,377 | | |
| - | | |
| - | | |
| 2,788,650 | |
| Exercise of pre-funded warrants | |
| - | | |
| - | | |
| 191,431 | | |
| 173,123 | | |
| - | | |
| - | | |
| - | | |
| 173,123 | |
| Equity issuance costs | |
| | | |
| | | |
| | | |
| | | |
| (386,332 | ) | |
| | | |
| | | |
| (386,332 | ) |
| Stock based compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| 125,644 | | |
| - | | |
| - | | |
| 125,644 | |
| Defined pension plan adjustments | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 266,924 | | |
| 266,924 | |
| Deemed dividend | |
| - | | |
| - | | |
| - | | |
| - | | |
| 2,076,180 | | |
| (2,076,180 | ) | |
| - | | |
| - | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (1,980,810 | ) | |
| - | | |
| (1,980,810 | ) |
| BALANCE, DECEMBER 31, 2024 | |
| 206,452 | | |
$ | 166,353 | | |
| 3,159,535 | | |
$ | 2,740,958 | | |
$ | 72,820,671 | | |
$ | (74,430,474 | ) | |
$ | 108,853 | | |
$ | 1,406,361 | |
Adjusted for the effect of a 1-to-40 reverse share split that was effective
September 27, 2024 (See note 2).
The accompanying notes are an integral part of
these consolidated financial statements.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | |
For the Years Ended December 31, | |
| | |
2024 | | |
2023 | | |
2022 | |
| Cash Flows From Operating Activities: | |
| | |
| | |
| |
| Net loss | |
$ | (1,980,810 | ) | |
$ | (12,172,029 | ) | |
$ | (16,495,680 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | |
| | | |
| | | |
| | |
| Periodic pension costs | |
| 6,239 | | |
| 59,526 | | |
| (34,142 | ) |
| Depreciation expense | |
| 3,321 | | |
| 11,408 | | |
| 11,408 | |
| Stock-based compensation | |
| 125,644 | | |
| 164,906 | | |
| 22,730 | |
| Other income due to the termination of EF License Agreement | |
| (2,499,969 | ) | |
| - | | |
| - | |
| Amortization of debt discounts | |
| - | | |
| - | | |
| 67,008 | |
| Loss on conversion of convertible notes payable | |
| - | | |
| - | | |
| 922,495 | |
| Changes in operating assets and liabilities: | |
| | | |
| | | |
| | |
| Prepaid expenses and other current assets | |
| 365,225 | | |
| (632,125 | ) | |
| 698,116 | |
| Other assets | |
| 16,602 | | |
| | | |
| | |
| Accounts payable | |
| (118,047 | ) | |
| 2,260,258 | | |
| 636,263 | |
| Interest payable | |
| - | | |
| 25,605 | | |
| 20,121 | |
| Other accrued liabilities | |
| (186,089 | ) | |
| 640,228 | | |
| 319,989 | |
| Other long term liabilities | |
| - | | |
| (42,243 | ) | |
| (47,679 | ) |
| Net cash used in operating activities | |
| (4,267,884 | ) | |
| (9,684,466 | ) | |
| (13,879,371 | ) |
| | |
| | | |
| | | |
| | |
| Cash Flows From Investing Activities: | |
| | | |
| | | |
| | |
| Purchases of property and equipment | |
| (3,917 | ) | |
| - | | |
| - | |
| Net cash used in investing activities | |
| (3,917 | ) | |
| - | | |
| - | |
| | |
| | | |
| | | |
| | |
| Cash Flows From Financing Activities: | |
| | | |
| | | |
| | |
| Proceeds from the issuance of common shares in ATM financing | |
| - | | |
| - | | |
| 30,993 | |
| Proceeds from the issuance of common shares in registered direct offerings | |
| - | | |
| - | | |
| 1,911,513 | |
| Proceeds from the issuance of common shares in private placement offerings | |
| 5,252,725 | | |
| - | | |
| 7,275,549 | |
| Proceeds from the issuance of pre-funded warrants | |
| - | | |
| - | | |
| 5,348,882 | |
| Proceeds from issuance of convertible notes payable | |
| - | | |
| - | | |
| 1,530,000 | |
| Exercise of pre-funded warrants | |
| 173,123 | | |
| - | | |
| 23,692 | |
| Equity issuance costs | |
| (386,332 | ) | |
| - | | |
| (704,160 | ) |
| Proceeds from the issuance of warrants | |
| - | | |
| - | | |
| 1,980,200 | |
| Proceeds from related party short term loan | |
| - | | |
| 1,633,746 | | |
| - | |
| Net cash provided by financing activities | |
| 5,039,516 | | |
| 1,633,746 | | |
| 17,396,669 | |
| | |
| | | |
| | | |
| | |
| Effect of exchange rate on cash and cash equivalents | |
| - | | |
| - | | |
| (100 | ) |
| | |
| | | |
| | | |
| | |
| Change in cash and cash equivalents | |
| 767,715 | | |
| (8,050,720 | ) | |
| 3,517,198 | |
| Cash and cash equivalents at the beginning of period | |
| 897,680 | | |
| 8,948,400 | | |
| 5,431,202 | |
| Cash and cash equivalents at the end of period | |
$ | 1,665,395 | | |
$ | 897,680 | | |
$ | 8,948,400 | |
| | |
| | | |
| | | |
| | |
| Supplemental disclosure of non-cash investing and financing activities: | |
| | | |
| | | |
| | |
| Cash paid for interest | |
$ | 467 | | |
$ | 145,153 | | |
$ | 13,248 | |
| Issuance of note payable for prepaid insurance | |
$ | - | | |
$ | | | |
$ | 704,160 | |
| Issuance of preferred shares for the assumption of debt | |
$ | 4,000,000 | | |
$ | - | | |
$ | - | |
| Issuance of common shares for debt conversion | |
| 2,788,650 | | |
| | | |
| | |
| Issuance of common shares for conversion of preferred shares | |
| 18,000 | | |
| | | |
| | |
Change in accrued pension liability reflected in accumulated other
comprehensive income | |
| 260,685 | | |
| | | |
| | |
| Deemed dividend | |
| 2,076,180 | | |
$ | - | | |
$ | - | |
| Debt discount on convertible loans | |
$ | - | | |
$ | - | | |
$ | 67,008 | |
The accompanying notes are an integral part of
these consolidated financial statements.
Note 1
Background:
NLS Pharmaceutics Ltd. (Nasdaq: NLSP, NLSPW) (the
“Company”) and its wholly-owned subsidiaries NLS Pharmaceutics (Israel) Ltd., an Israeli company (the “Merger Sub”)
and NLS Pharmaceutics Inc., a Delaware corporation, is an emerging biopharmaceutical company engaged in the discovery and development
of life-improving drug therapies to treat rare and complex central nervous system disorders, including narcolepsy, idiopathic hypersomnia
and other rare sleep disorders, and of neurodevelopmental disorders, such as attention deficit hyperactivity disorder (“ADHD”).
The Company’s lead product candidates are Quilience, to treat narcolepsy (type 1 and type 2), and Nolazol, to treat ADHD.
The Company was established in June 2015 through
the incorporation of its predecessor entities, NLS Pharma Ltd. and NLS-1 Pharma Ltd., in Switzerland. In April 2016, NLS-0 Pharma Ltd.
was also incorporated. Subsequently, in a reorganization effective January 1, 2019, NLS Pharma Ltd. and NLS-0 Pharma Ltd. merged into
NLS-1 Pharma Ltd., which was then renamed NLS Pharmaceutics Ltd. The Company went public on January 29, 2021, with its shares listed on
the Nasdaq Capital Market. On April 27, 2021, the Company formed NLS Pharmaceutics Inc., a Delaware corporation (“NLS Inc.”).
On November 3, 2024, the Company formed NLS Pharmaceutics (Israel) Ltd., an Israeli company for the purposes of the potential merger transaction.
The accompanying consolidated financial statements include the results
of the Company and its wholly-owned subsidiaries. All references hereinafter to the Company mean the Company and its subsidiaries
NLS Inc. and the Merger Sub.
Agreement and Plan of Merger
On November 4, 2024, the Company, the Merger Sub,
and Kadimastem Ltd., an Israeli publicly traded company limited by shares (TASE: KDST) (“Kadimastem”), entered into an Agreement
and Plan of Merger (the “Merger Agreement”), pursuant to which (i) Kadimastem will merge with and into Merger Sub, with Merger
Sub as the surviving company (the “Merger”), and (ii) at the effective time of the Merger (the “Effective Time”),
each issued and outstanding ordinary share of Kadimastem, no par value (“Kadimastem Ordinary Share”), will be exchanged for
and automatically converted into the right to receive from the Company that certain number of fully paid and nonassessable common shares,
0.03 Swiss Franc (CHF) par value per share, of the Company (“Company Common Share”) as calculated in accordance with the terms
of the Merger Agreement (the “Exchange Ratio”). It is anticipated that the initial Exchange Ratio is estimated to result in
Kadimastem shareholders holding 80% of the issued and outstanding shares of Company Common Shares on a fully diluted basis, subject to
certain adjustments as of the closing of the Merger (the “Closing”).
The Merger Agreement provides that, upon the terms
and subject to the conditions thereof, following the Closing, the Company shall work diligently to dispose of any intellectual property,
assets, rights, contracts, agreements, leases, arrangements (regardless of form), approvals, licenses, permits, whether current or future,
whether or not contingent, of the Company and its subsidiaries related solely to any product candidate of the Company and its subsidiaries,
other than the Company’s Dual Orexin Agonist platform (such assets to be disposed, the “Legacy Assets”). It is expected
that the proceeds from any such disposition will be distributed to the shareholders and warrant holders of the Company as of immediately
prior to the Effective Time pursuant to the terms and conditions of a contingent value rights agreement, (the “CVR Agreement”).
At the Effective Time, each:
| ● | Kadimastem Ordinary Share issued and outstanding immediately prior to the Effective Time will be exchanged
for and converted into the right to receive a number of newly issued, fully paid and nonassessable Company Common Shares equal to the
Exchange Ratio; |
| ● | option, restricted share unit, restricted share, warrant or other rights issued and outstanding, whether
vested or unvested, to purchase Kadimastem Ordinary Shares, shall be assumed by the Company and converted into an option, warrant, other
award, or right, as applicable, to purchase Company Common Shares in accordance with the terms of the Merger Agreement: |
| ● | each Company Common Share issued and outstanding immediately prior to the Effective time, and each Company
Common Share acquirable upon the exercise of outstanding warrants and pre-funded warrants of the Company, shall continue to remain outstanding
and, in addition, be entitled to a contingent value right (“CVR”) pursuant to the terms of the Merger Agreement and the CVR
Agreement. |
The Merger Agreement and the consummation of the
transactions contemplated thereby have been approved by the Company’s board of directors (the “Board”) and Kadimastem’s
board of directors, and the Board has resolved, subject to customary exceptions, to recommend that the shareholders of the Company approve
the Merger Agreement and the transactions contemplated therein.
The Merger Agreement contains customary termination
rights for each of the Company and Kadimastem, including the right of the Company and Kadimastem to terminate the Merger Agreement if
the Closing shall not have occurred on or before June 30, 2025. The Merger Agreement also provides that the Company shall pay to Kadimastem
a termination fee of $10.0 million plus the Company Operating Expenses (as defined in the Merger Agreement), and the Transaction Expenses
(as defined in the Merger Agreement) if the Company terminates the Merger Agreement prior to obtaining the Parent Requisite Vote (as defined
in the Merger Agreement) to enter into a definitive agreement providing for a Parent Superior Proposal (as defined in the Merger Agreement)
in accordance with terms of the Merger Agreement.
Contingent Value Right Agreement
Prior to the Closing, the Company will enter into
the CVR Agreement with VStock Transfer, LLC, which will govern the terms of the CVRs. Each CVR will represent the right to additional
payments based on the proceeds, subject to certain adjustments, received by the Company from the disposition of the Legacy Assets.
The right to the CVRs as evidenced by the CVR
Agreement is a contractual right only and will not be transferable, except in the limited circumstances specified in the CVR Agreement.
Going Concern
As of December 31, 2024, the Company had an accumulated
deficit of approximately $74.4 million and the Company incurred an operating loss for the year ended December 31, 2024, of approximately
$4.4 million. To date, the Company has dedicated most of its financial resources to achieve and maintain Phase 3 readiness, research and
development, clinical studies associated with its ongoing biopharmaceutical business and general and administrative expenses. The Company
has not generated revenues from its activities and has incurred substantial operating losses.
As of December 31, 2024, the Company’s
cash and cash equivalents were $1.7 million. The Company’s existing cash and cash equivalents and access to existing financing
arrangements will not be sufficient to fund operations for a period of one year from the issuance of these consolidated financial
statements. The Company expects to continue to generate operating losses and negative operating cash flows for the next few years
and will need additional funding to support its planned operating activities through profitability. The Company is actively
exploring a range of options to raise funds, including strategic partnerships, out-licensing, or divestment of assets of the
Company, and other future strategic actions. During October 2024, the Company completed a private financing round, debt conversions
and forgiveness, and various vendor buy-outs. Additionally, on November 4, 2024, the Company entered into the Merger Agreement. The
future viability of the Company is dependent on its ability to raise capital for payment of its vendors and to support on-going
operations. There can be no assurance that such capital will be available within a sufficient period of time, in sufficient amounts
or on terms acceptable to the Company. These conditions raise substantial doubt about the Company’s ability to continue as a
going concern beyond one year from the issuance of these consolidated financial statements.
Accordingly, the accompanying consolidated financial
statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S.
GAAP”), which contemplate continuation of the Company as a going concern for a period within one year from the issuance of these
consolidated financial statements and the realization of assets and satisfaction of liabilities in the normal course of business. The
carrying amounts of assets and liabilities presented in these consolidated financial statements do not necessarily purport to represent
realizable or settlement values. These consolidated financial statements do not include any adjustment that might result from the outcome
of this uncertainty.
Note 2
Summary of Significant Accounting Policies:
Basis of Presentation
The accompanying consolidated financial statements
have been prepared in accordance with U.S. GAAP. Any reference in these notes to applicable guidance is meant to refer to the authoritative
U.S. GAAP as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Updates (“ASU”) of
the Financial Accounting Standards Board (“FASB”).
Effective September 27, 2024,
the Company filed amended Articles of Association with the commercial registry of Zurich reflecting an increase in share capital to CHF
937,600, divided into 1,172,000 registered shares with a nominal value of CHF 0.80 each and filed for a 1-for-40 reverse share split.
The number of shares outstanding before and after the reverse split were adjusted accordingly on a retrospective basis. Further, on January
14, 2025, the shareholders of NLS approved a change in the par value of the common share from CHF 0.80 to CHF 0.03 per share, effective
January 17, 2025. All share amounts reflect the par value of CHF 0.80 as of December 31, 2024 and no adjustments have been made for the
change in par value to additional paid in capital for the reduction in par value.
Use of Estimates
The preparation of the consolidated financial
statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect amounts reported of assets and
liabilities at the date of the consolidated financial statements and the reported amounts of expenses during the reporting periods. Actual
results could differ from those estimates and be based on events different from those assumptions. As part of these consolidated financial
statements, the Company’s significant estimates include stock-based compensation, pension liabilities and the valuation allowance
related to the Company’s deferred tax assets.
JOBS Act Accounting Election
The Company is an “emerging growth company”
(“EGC”) as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”). Under the JOBS Act, an
EGC can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those
standards apply to private companies. The Company intends to take advantage of the exemptions until it is no longer an EGC.
Cash and Cash Equivalents
The Company considers all highly liquid
investments with an original maturity of three months or less at the date of purchase to be cash equivalents. The Company deposits
its cash primarily in checking, money market accounts, as well as certificates of deposit. The Company generally does not enter into
investments for trading or speculative purposes rather to preserve its capital for the purpose of funding operations.
Property and equipment
Property and equipment are recorded at cost, net
of accumulated depreciation and any accumulated impairment losses. Depreciation is computed using the straight-line method over the estimated
useful lives of the assets. The useful lives of property and equipment are five years for furniture and fixtures and three years for software.
Upon retirement or sale, the cost of disposed
assets and their related accumulated depreciation are removed from the balance sheet. Any resulting net gains or losses on dispositions
of property and equipment are included as a component of operating expenses within the Company’s statements of operating and comprehensive
loss. Repair and maintenance costs that do not significantly add value to the property and equipment, or prolong its life, are charged
to operating expense as incurred.
Concentration of Credit Risk
Financial instruments that potentially
subject the Company to concentration of credit risk include cash. On December 31, 2024 and 2023, all of the Company’s cash
balances are deposited in one banking institution in Switzerland. At various times, the Company has deposits in financial
institutions which are in excess of federally insured limits of CHF 100,000 ($109,650) that are protected by esisuisse. Management
regularly monitors the creditworthiness and financial stability of these banking institutions to mitigate credit risk. This review
includes periodic evaluations of the banks’ financial health, regulatory standings, and credit ratings.
Functional Currency
The Company has operations in Switzerland and
the United States. The Company’s functional currency is the U.S. dollar (“USD”). The results of its non-USD based operations
are translated to USD at the average exchange rates during the year. The Company’s assets and liabilities are translated using the
current exchange rate as of the balance sheet date and shareholders’ equity is translated using historical rates. Foreign exchange
transaction gains and losses are included in Other income/expense in the Company’s results of operations.
Revenue Recognition
Under ASC Topic 606 and the related amendments, Revenue from Contracts
with Customers, the Company recognizes revenue when control of promised goods or services is transferred to the customer. The five-step
model is applied, which includes identifying performance obligations, determining the transaction price, allocating the transaction price,
and recognizing revenue when obligations are satisfied. Performance obligations are distinct when the customer can benefit from them independently,
and the Company evaluates the distinctiveness considering factors such as intellectual property development and customer capabilities.
Variable consideration is estimated based on the
likelihood of achievement, with constrained amounts included only if a significant reversal is unlikely. Transaction prices are allocated
to performance obligations based on stand-alone selling prices, which may require judgment and references to comparable transactions,
clinical trial probabilities, and expected option exercises.
Revenue from development and regulatory milestones
is recognized under the most likely amount method, subject to the constraint that revenue is only recognized when it is probable that
a significant reversal will not occur. Milestones tied to regulatory approvals or other external conditions are not considered probable
until those conditions are met. At each reporting period, the Company reassesses the likelihood of milestone achievement and adjusts the
transaction price as necessary. Any adjustments are recorded on a cumulative catch-up basis, impacting license revenues in the period
of adjustment.
Sales-based royalties are recognized when the
related sales occur or when the performance obligation is satisfied. Deferred revenue is recorded when payments exceed recognized revenue,
including non-refundable payments.
As of December 31, 2024, the Company recognized
$2.5 million from its exclusive license agreement (the “EF License Agreement”) as Other income due to the termination of the
EF License Agreement. The amount represents a non-refundable upfront payment, which was retained by the Company after the early termination
of the EF License Agreement, and falls outside the scope of ASC 606. See Note 7 for further details.
Research and Development
Costs for R&D of products, including vendor
expenses and supplies and consultant fees, are expensed as incurred. Clinical trial and other development costs incurred by third parties
are expensed as the contracted work is performed. Where contingent milestone payments are due to third parties under research and development
arrangements, the obligations are recorded when the milestone results are probable of being achieved.
Fair Value Measurements
The Company measures and discloses fair value
in accordance with ASC 820, “Fair Value,” which defines fair value, establishes a framework and gives guidance regarding
the methods used for measuring fair value, and expands disclosures about fair value measurements. Fair value is an exit price, representing
the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants
at the measurement date. As such, fair value is a market-based measurement that should be determined based on assumptions that market
participants would use in pricing an asset or liability. As a basis for considering such assumptions there exists a three-tier fair-value
hierarchy, which prioritizes the inputs used in measuring fair value as follows:
Level 1 - unadjusted quoted prices are
available in active markets for identical assets or liabilities that the Company has the ability to access as of the measurement date.
Level 2 - pricing inputs are other than
quoted prices in active markets that are directly observable for the asset or liability or indirectly observable through corroboration
with observable market data.
Level 3 - pricing inputs are
unobservable for the non-financial asset or liability and only used when there is little, if any, market activity for the
non-financial asset or liability at the measurement date. The inputs into the determination of fair value require significant
management judgment or estimation. Fair value is determined using comparable market transactions and other valuation methodologies,
adjusted as appropriate for liquidity, credit, market and/or other risk factors.
This hierarchy requires the Company to use observable
market data, when available, and to minimize the use of unobservable inputs when determining fair value.
The Company’s cash and cash equivalents
are carried at fair value, determined according to the fair value hierarchy described above. The carrying value of the Company’s
accounts payable and accruals approximates its fair value due to the short-term nature of these liabilities.
Debt Issuance Costs and Debt Discount
Debt issuance costs related to a recognized debt liability are presented
on the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts, and are amortized
to interest expense over the term of the related debt using the effective interest method. As of December 31, 2024 and 2023, the Company
had no unamortized debt discount associated with its outstanding debt instruments.
Income Taxes
The Company accounts for income taxes using the
asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences
of events that have been recognized in the consolidated financial statements or in the Company’s tax returns. Deferred taxes are
determined based on the difference between the consolidated financial statement and tax basis of assets and liabilities using enacted
tax rates in effect in the years in which the differences are expected to reverse. Changes in deferred tax assets and liabilities are
recorded in the provision for income taxes. The Company assesses the likelihood that its deferred tax assets will be recovered from future
taxable income and, to the extent it believes, based upon the weight of available evidence, that it is more likely than not that all or
a portion of the deferred tax assets will not be realized, a valuation allowance is established through a charge to income tax expense.
Potential for recovery of deferred tax assets is evaluated by estimating the future taxable profits expected and considering prudent and
feasible tax planning strategies.
Due to the fact that the Company has a history
of generating losses, and expects to generate losses in the foreseeable future, a full valuation allowance has been recorded.
The Company accounts for uncertain tax positions
in accordance with an amendment to ASC Topic 740-10, “Income Taxes (Accounting for Uncertainty in Income Taxes),”
which clarified the accounting for uncertainty in tax positions. This amendment provides that the tax effects from an uncertain tax
position can be recognized in the consolidated financial statements only if the position is “more-likely-than-not” to be sustained
were it to be challenged by a taxing authority. The assessment of the tax position is based solely on the technical merits of the position,
without regard to the likelihood that the tax position may be challenged. If an uncertain tax position meets the “more-likely-than-not”
threshold, the largest amount of tax benefit that is more than 50% likely to be recognized upon ultimate settlement with the taxing authority
is recorded.
Employee Benefits (including Post Retirement Benefits)
The Company operates the mandatory pension plan
for its employees in Switzerland. The plan is generally funded through payments to insurance companies or trustee-administered funds.
The Company has a pension plan designed to pay pensions based on accumulated contributions on individual savings accounts. However, this
plan is classified as a defined benefit plan under ASC 960 “Plan Accounting – Defined Benefit Pension Plans.”
The net defined benefit liability is the present
value of the defined benefit obligation at the balance sheet date minus the fair value of plan assets. The defined benefit obligation
is calculated annually by independent actuaries using the projected unit credit method, which reflects services rendered by employees
to the date of valuation, incorporates assumptions concerning employees’ projected salaries, pension increases as well as discount
rates of highly liquid corporate bonds which have terms to maturity approximating the terms of the related liability.
Remeasurements of the net defined benefit liability,
which comprise actuarial gains and losses, and the return on plan assets (excluding interest), are recognized immediately in Other Comprehensive
Loss. Past service costs, including curtailment gains or losses, are recognized immediately as an allocation between research and development
and general and administrative expenses within the operating results. Settlement gains or losses are recognized in either research and
development and/or general and administrative expenses within the operating results. The Company determines the net interest expense (income)
on the net defined benefit liability for the period by applying the discount rate used to measure the defined benefit obligation at the
beginning of the annual period or in case of any significant events between measurement dates to the then-net defined benefit liability,
taking into account any changes in the net defined benefit liability during the period as a result of contributions and benefit payments.
Net interest expense and other expenses related to defined benefit plans are recognized in the statement of operations and comprehensive
loss.
Stock-Based Compensation
The Company measures all stock-based awards granted
based on the fair value on the date of the grant and recognizes compensation expense with respect to those awards over the requisite service
period, which is generally the vesting period of the respective award. Generally, the Company issues awards with only service-based vesting
conditions and records the expense for these awards using the straight-line method. The Company recognizes forfeitures related to stock-based
compensation awards as they occur and reverses any previously recognized compensation cost associated with forfeited awards in the period
the forfeiture occurs.
The Company classifies stock-based compensation
expense in the accompanying consolidated statements of operations and comprehensive loss in the same manner in which the award recipients’
payroll costs are classified or in which the award recipients’ service payments are classified.
The fair value of each stock option is estimated
on the date of grant using the Black-Scholes option-pricing model (“Black-Scholes”). Black-Scholes requires a number of assumptions,
of which the most significant are share price, expected volatility, expected option term (the time from the grant date until the options
are exercised or expire), risk-free rate and expected dividend rate. The grant date fair value of a common share is determined by the
board of directors (the “Board of Directors”) considering, among other factors, the assistance of a valuation specialist and
management. The grant date fair value of a common share is determined using the valuation methodologies, which utilize certain assumptions,
including probability weighting of events, volatility, time to liquidation, risk-free interest rate and discount for lack of marketability.
Preferred Shares
Upon issuance of a convertible preferred share
instrument, the Company evaluates its classification as either equity or debt. In accordance with ASC 480, the Company’s
preferred shares were classified as permanent equity as it does not contain any mandatorily redeemable provisions. Further, in accordance
with ASC 815-40, Derivatives and Hedging – Contracts in an Entity’s Own Equity, the preferred shares did not meet any of
the criteria that would preclude equity classification. The Company concluded that the preferred shares were more akin to an equity-type
instrument than a debt-type instrument, therefore the conversion features associated with the convertible preferred shares were deemed
to be clearly and closely related to the host instrument and were not bifurcated as a derivative under ASC 815.
Earnings per Share
Basic net loss per common share is computed by
dividing the net loss applicable to common shareholders by the weighted-average number of common shares outstanding during the year.
Diluted loss per common share is computed similar to basic loss per share, except that the denominator is increased to include the number
of additional potential common shares that would have been outstanding if the potential common shares had been issued and if the additional
common shares were dilutive. Potential common shares are excluded from the diluted loss per common share computation for a period in
which a net loss is reported or if their effect is anti-dilutive. Since the Company was in a loss position for all periods presented,
basic and diluted weighted average shares outstanding are the same, as the inclusion of the incremental shares would be anti-dilutive.
A summary of the potentially dilutive securities that were excluded from diluted net loss per share each year because their effect would be antidilutive are presented as follows:
| | |
December 31, | |
| | |
2024 | | |
2023 | | |
2022 | |
| Stock Options | |
| 34,417 | | |
| 46,591 | | |
| 33,328 | |
| Warrants (excluding pre-funded) | |
| 1,455,288 | | |
| 332,450 | | |
| 332,450 | |
| Convertible preferred shares | |
| 206,452 | | |
| - | | |
| - | |
| | |
| 1,696,157 | | |
| 379,041 | | |
| 365,778 | |
Adjusted for the effect of a 1-to-40 reverse share split that was effective
September 27, 2024
Segment Reporting
The Company manages its operations as a single
segment for the purpose of assessing performance and making operating decisions. The Company’s singular focus is on developing therapeutics
for the treatment of neurobehavioral and neurocognitive disorders. All of the Company’s tangible assets are held in Switzerland.
Operating segments are defined as components of an entity for which separate financial information is available and that is regularly
provided to the Chief Operating Decision Maker (“CODM”) in deciding how to allocate resources to an individual segment and
in assessing performance. The Company’s Chief Executive Officer is the Company’s CODM. The CODM reviews financial information
presented on a consolidated basis for purposes of making operating decisions, allocating resources, and evaluating financial performance.
The CODM uses consolidated net income (loss) to assess performance, evaluate cost optimization, and allocate resources, including personnel-related
and financial or capital resources, in the annual budget and forecasting process, as well as budget-to-actual variances on a monthly basis.
As such, the Company has determined that it operates as one operating and reportable segment.
The significant expenses regularly reviewed by
the CODM are consistent with those reported on the Company’s consolidated statement of operations and expenses are not regularly
reviewed on a more disaggregated basis for assessing segment performance and deciding how to allocate resources. The CODM does not regularly
review total assets for the Company’s single reportable segment as total assets are not used to assess performance or allocate resources.
Recently Adopted Accounting Pronouncements
In November 2023, the Financial Accounting Standards
Board (FASB) issued Accounting Standards Update (“ASU”) No. 2023-07, Improvements to Reportable Segment Disclosures (Topic
280). This ASU enhances segment disclosure requirements by requiring entities to disclose significant segment expenses that are regularly
provided to the CODM and included in each reported measure of a segment’s profit or loss. Additionally, the ASU requires disclosure
of the title and position of the individual identified as the CODM, along with a description of how the CODM uses the reported segment
measures to assess performance and allocate resources. For the Company, the Chief Executive Officer (CEO) serves as the CODM, as the
CEO is responsible for allocating resources to and assessing the performance of the Company’s operating segments.
The standard is effective for annual periods
beginning after December 15, 2023, and for interim periods within fiscal years beginning after December 15, 2024. The Company adopted
the ASU retrospectively as of December 31, 2024. The adoption of this standard had an immaterial impact on the Company’s
consolidated financial statements and related disclosures for all periods presented.
Recent Accounting Pronouncements Not Yet
Adopted
In December 2023, FASB issued Accounting Standards Update (ASU) 2023-09, Income Taxes
(Topic 740): Improvements to Income Tax Disclosures (ASU 2023-09). The ASU focuses on income tax disclosures around effective tax rates
and cash income taxes paid. ASU 2023-09 requires public business entities to disclose, on an annual basis, a rate reconciliation presented
in both dollars and percentages. The guidance requires the rate reconciliation to include specific categories and provides further guidance
on disaggregation of those categories based on a quantitative threshold equal to 5% or more of the amount determined by multiplying pretax
income (loss) from continuing operations by the applicable statutory rate. For entities reconciling to the US statutory rate of 21%, this
would generally require disclosing any reconciling items that impact the rate by 1.05% or more. ASU 2023-09 is effective for public business
entities for annual periods beginning after December 15, 2024 (generally, calendar year 2025) and effective for all other business entities
one year later. Entities should adopt this guidance on a prospective basis, though retrospective application is permitted. The adoption
of ASU 2023-09 is expected to have a financial statement disclosure impact only and is not expected to have a material impact on the Company’s
consolidated financial statements.
In November 2024, the FASB issued ASU 2024-03, Disaggregation
of Income Statement Expenses (“DISE”), a new accounting standard to improve the disclosures about an entity’s expenses
and address requests from investors for more detailed information about the types of expenses included in commonly presented expense captions.
The new standard is effective for annual reporting periods beginning after December 15, 2026, and interim reporting periods beginning
after December 15, 2027, with retrospective application permitted. The Company is evaluating the disclosure requirements related to the
new standard and its impact on the Company’s consolidated financial statements.
The Company has implemented all new
accounting pronouncements currently in effect that may impact its consolidated financial statements and does not believe any
other recently issued standards will have a material impact on its financial position or results of operations.
Note 3
Prepaid Expenses and Other Current Assets:
The Company’s prepaid expenses and other
current assets consisted of the following as of December 31, 2024, and 2023:
| | |
December 31, | |
| | |
2024 | | |
2023 | |
| | |
| | |
| |
| Vendor prepayments | |
$ | 65,237 | | |
$ | 821,266 | |
| VAT recoverable and other current assets | |
| 49,479 | | |
| 24,433 | |
| Other short-term receivables | |
| 400,000 | | |
| 10,664 | |
| Prepaid insurance | |
| 42,470 | | |
| 69,019 | |
| Prepaid expenses | |
| 2,970 | | |
| - | |
| | |
| | | |
| | |
| Total prepaid expenses and other current assets | |
$ | 560,157 | | |
$ | 925,382 | |
During the period ended December 31, 2024, the
Company provided working capital advances totaling $400,000 to Kadimastem, a third party with which the Company is engaged in merger
negotiations. These funds were provided to support Kadimastem’s ongoing operations and are recorded as Other Receivable on the
balance sheet. The funds are repayable on demand or upon termination of the merger discussions. If the merger is completed, the advances
will be eliminated in consolidation. The Company’s ability to recover these funds is dependent on the outcome of the merger transaction.
As of December 31, 2024, no allowance for uncollectibility has been recorded, as management believes the amounts are recoverable.
Note 4
Property and Equipment, net:
The Company’s property and equipment consisted
of the following as of December 31, 2024 and 2023:
| | |
December 31, | |
| | |
2024 | | |
2023 | |
| Cost | |
| | |
| |
| Furniture and fixtures | |
$ | 17,258 | | |
$ | 13,341 | |
| Software | |
| 26,219 | | |
| 26,219 | |
| Total cost | |
| 43,477 | | |
| 39,560 | |
| Accumulated depreciation | |
| (36,187 | ) | |
| (32,866 | ) |
| | |
| | | |
| | |
| Total property and equipment, net | |
$ | 7,290 | | |
$ | 6,694 | |
Depreciation expense was $3,321, $11,408 and $11,408
for the years ended December 31, 2024, 2023 and 2022.
Note 5
Related party short-term loans:
On September 28, 2023 and November 15, 2023, the
Company entered into a series of short term loan agreements (the “Short Term Loan Agreements”) with certain existing shareholders
of the Company, including Ronald Hafner, the Company’s Chairman of the Board of Directors, Felix Grisard, Jürgen Bauer and
Maria Nayvalt, providing for unsecured loans to the Company in the aggregate amount of CHF 1,375,000 ($1,510,989). The loans bear interest
at a rate of 10% per annum and mature on the earlier of December 31, 2024, or a liquidity event with a strategic partner, as amended on
March 18, 2024.
In October 2024, the Company entered into debt
forgiveness agreements with the holders of the related party short-term loans, pursuant to which the Company issued an aggregate of 290,518
common shares, par value CHF 0.03 per share, or the common shares, of the Company, at a conversion price of $5.65 (rounded) per share,
in full satisfaction of an aggregate amount of CHF 1,492,431 ($1,640,034) in loans and interest provided to the Company by the Lenders,
or collectively, the Loans. This conversion was facilitated through an ordinary capital increase, providing the necessary shares for the debt holders, and was recorded
at fair market value with no gain or loss recognized in the statement of operations.
Note 6
Other Accrued Liabilities:
The Company’s other accrued liabilities
consisted of the following as of December 31, 2024 and 2023:
| | |
December 31, | |
| | |
2024 | | |
2023 | |
| | |
| | |
| |
| Professional consultants’ expenses | |
$ | 110,345 | | |
$ | 332,690 | |
| Vendor liabilities | |
| - | | |
| 112,635 | |
| Stamp tax | |
| 125,793 | | |
| - | |
| Related party interest short term loan | |
| - | | |
| 25,605 | |
| Accrued Board of Directors fees | |
| 47,586 | | |
| 184,672 | |
| Accrued bonus | |
| - | | |
| 960,025 | |
| Other accrued expenses | |
| 27,554 | | |
| 36,643 | |
| | |
| | | |
| | |
| Total other accrued liabilities | |
$ | 311,278 | | |
$ | 1,652,270 | |
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
Note 7
Deferred Revenues:
In February 2019, the Company entered into the
EF License Agreement, to develop and commercialize its product candidate, Nolazol, in Latin American countries with Eurofarma Laboratorios
S.A., a Brazilian pharmaceutical company. The EF License Agreement covered the grant of non-transferable licenses, without the right to
sublicense, to Eurofarma to develop and commercialize Nolazol in Latin America. The EF License Agreement also specified the Company’s
obligation to advance ongoing development activities with respect to Nolazol in the United States. A joint steering committee oversaw
the development and regulatory activities directed towards marketing approval, manufacturing and commercialization phases. The Company
believed its participation in the joint steering committee was not of material significance to the licenses in the context of the EF License
Agreement on the whole and, as such, management excluded these activities in the determination of its performance obligation(s) under
the EF License Agreement.
The EF License Agreement provided that the parties
would enter into a separate manufacturing and supply agreement during the term of the EF License Agreement.
Under the EF License Agreement, the Company received
a non-refundable, upfront payment, of $2.5 million and was further eligible to receive non-refundable milestone payments of up to $16
million, based on the achievement of milestones related to regulatory filings, regulatory approvals and the commercialization of Nolazol.
The achievement and timing of the milestones depended on the success of development, approval and sales progress, if any, of Nolazol in
the future. In addition, the Company was also eligible for tiered royalty payments.
The Company identified the licenses granted to
Eurofarma and its obligation to advance development activities with respect to Nolazol in the United States as the material promises under
the EF License Agreement. For purposes of identifying the Company’s performance obligations under the EF License Agreement, management
believed that while the exclusive licenses were granted to Eurofarma at the outset of the EF License Agreement, the grant of those licenses
did not singularly result in the transfer of the Company’s broader obligation to Eurofarma under the EF License Agreement.
The Company was obligated under the EF License
Agreement to advance its development activities in the United States and those activities preceded Eurofarma’s necessary regulatory
approvals for commercialization of Nolazol, in Latin American countries. The Company intended to apply its proprietary know-how to the
ongoing development activities in the United States involving its intellectual property relating to Nolazol. These development activities
were specific to the Company and the Company believed they were not capable of being distinct in the context of the EF License Agreement
on the whole.
The licenses provided to Eurofarma were not
transferable and without the right to sublicense therefore Eurofarma was not presently able to monetize its investment in Nolazol as
clinical development in the United States or any Latin American countries had yet to be completed and Eurofarma had yet to seek or
obtain regulatory approval in any Latin American country. The licenses to Eurofarma represented rights to use the Company’s
intellectual property with respect to Nolazol for which revenue was recognized at a point in time which was when Eurofarma was able
to use and benefit from the licenses. The licenses were considered of limited value without the Company’s development
activities with respect to Nolazol in the United States. As such, the licenses were not capable of being distinct until after
successful clinal development and regulatory approval and alone did not have standalone functionality to Eurofarma. Management had
determined that the licenses, while capable of being distinct, were not distinct as they did not have stand-alone value to Eurofarma
without the Company’s planned development activities in the United States and the approval for sale in Latin America.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
Bundled together with the Company’s development
activities of Nolazol in the United States, the licenses granted under the EF License Agreement enabled Eurofarma to seek regulatory approvals
and ultimately seek to commercialize Nolazol in Latin America. Therefore, management believed the licenses bundled together with the Company’s
development activities in the United States constituted a single distinct performance obligation under the EF License Agreement for accounting
purposes (the “License Performance Obligation”).
The Company had initially estimated a total transaction
price of $2.5 million, consisting of the fixed upfront payment determined to be an advance on the License Performance Obligation. Upon
execution of the EF License Agreement, variable consideration consisting of milestone payments had been constrained and excluded from
the transaction price given the significant uncertainty of achievement of the development and regulatory milestones. As a result, the
Company allocated the transaction price entirely to a single License Performance Obligation and recorded $2.5 million as deferred revenue,
which would have been recognized when the development services of Nolazol were completed, and the product candidate received applicable
regulatory approval in Latin America that allowed Eurofarma to commence commercialization of Nolazol in accordance with the EF License
Agreement.
On August 28, 2024, the Company agreed with Eurofarma
to terminate the EF License Agreement effective September 30, 2024. Neither party has any claims against the other in relation to the
EF License Agreement and its termination. As of December 31, 2024, the Company recognized $2.5 million from its exclusive license agreement
(the “EF License Agreement”) in the Statements of Operations and Comprehensive loss as Other income for termination of EF
License Agreement.
Note 8
Pension Liability:
The Company joined a collective pension plan operated
by an insurance company as of 2016 which covers the employees in Switzerland.
Both the Company and the participants provide
monthly contributions to the pension plan which are based on the covered salary. The respective saving parts of premiums are credited
to employees’ accounts. In addition, interest is credited to employees’ accounts at the rate provided in the plan. The pension
plan provides for retirement benefits as well as benefits on long-term disability and death.
The following table provides information on the
pension plan for the years ended December 31, 2024, 2023 and 2022:
| | |
2024 | | |
2023 | | |
2022 | |
| Service cost | |
$ | 69,716 | | |
$ | 53,920 | | |
$ | 39,667 | |
| Interest cost | |
| 24,375 | | |
| 14,416 | | |
| 2,743 | |
| Expected return on assets | |
| (32,364 | ) | |
| (14,767 | ) | |
| (12,196 | ) |
| Effect of settlement, curtailment, plan amendment | |
| (44,236 | ) | |
| - | | |
| (72,600 | ) |
| Actuarial loss outside corridor recognized | |
| 5,231 | | |
| - | | |
| 3,091 | |
| Past service cost recognized in year | |
| 3,094 | | |
| 3,031 | | |
| 2,855 | |
| Administrative expenses | |
| 1,861 | | |
| 2,926 | | |
| 2,298 | |
| | |
| | | |
| | | |
| | |
| Net periodic pension cost | |
$ | 27,677 | | |
$ | 59,525 | | |
$ | (34,142 | ) |
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The reconciliation of the projected benefit obligation
and the changes to the fair value of the plan assets of the pension plan are shown in the following table:
| | |
2024 | | |
2023 | |
| Projected benefit obligation, beginning of period | |
$ | 809,028 | | |
$ | 584,737 | |
| Service cost and plan amendments | |
| 26,283 | | |
| 53,920 | |
| Interest cost | |
| 24,375 | | |
| 14,416 | |
| Employer contributions | |
| 14,777 | | |
| (3,759 | ) |
| Transfers-in and (-out), net | |
| 1,342,430 | | |
| 42,413 | |
| Actuarial (gain)/ loss | |
| 21,240 | | |
| 123,388 | |
| Currency conversion adjustments | |
| (24,956 | ) | |
| (6,087 | ) |
| Projected benefit obligation, end of period | |
$ | 2,213,177 | | |
$ | 809,028 | |
| | |
| | | |
| | |
| Plan assets, beginning of period | |
$ | 548,343 | | |
$ | 448,615 | |
| Actual return on plan assets | |
| 32,364 | | |
| 27,844 | |
| Employer contributions | |
| 33,300 | | |
| 38,737 | |
| Participant contributions | |
| 14,777 | | |
| 20,655 | |
| Actuarial (gain)/ loss | |
| 178,872 | | |
| - | |
| Transfers-in and (-out), net | |
| 1,445,453 | | |
| 17,999 | |
| Administration expenses | |
| (1,861 | ) | |
| (2,926 | ) |
| Currency conversion adjustments | |
| (38,071 | ) | |
| (2,581 | ) |
| Plan assets, end of period | |
$ | 2,213,177 | | |
$ | 548,343 | |
| | |
| | | |
| | |
| Accrued pension liability | |
$ | - | | |
$ | 260,685 | |
As of December 31, 2024, the Company does not have any active employees
participating in a defined benefit pension plan. However, the Company continues to maintain a capped pension obligation related to survivor
benefits payable to orphans of a former employee. This obligation is fully covered by existing plan assets and does not give rise to any
unfunded liability. All other obligations and liabilities associated with the Company’s prior pension plan have been fully settled
and transferred. As a result, the Company has no net pension-related liability or associated pension expense recognized on its balance
sheet as of December 31, 2024.
The pension assets are measured at fair value
and are invested in a collective pension foundation with pooled investments. Plan assets mainly consist of cash and cash equivalents,
equity funds, equity securities, corporate bonds, government bonds, and real estate funds classified as Level 1 and Level 2 under the
fair value hierarchy.
The Company records net gains/losses, consisting
of actuarial gains/losses, curtailment gains/losses and differences between expected and actual returns on plan assets, in other comprehensive
income/loss. Such net gains/losses are amortized to the statements of operations to the extent that they exceed 10% of the greater of
projected benefit obligations or pension assets. The Company further records prior service costs/credits from plan amendments in other
comprehensive income/loss in the period of the respective plan amendment and amortizes such amounts to the statement of operations over
the future service period of the plan participants. For the years ended December 31, 2024 and 2023, the amortization was $0. As of December
31, 2024, and 2023, the accumulated other comprehensive (income) loss includes unrecognized pension costs of $(108,853) and $158,073,
respectively.
The following table shows the components of unrecognized
pension cost in accumulated other comprehensive income/loss that have not yet been recognized as components of net periodic pension cost:
| | |
2024 | | |
2023 | |
| Net loss, beginning of period | |
$ | 158,073 | | |
$ | 50,791 | |
| Other (gain)/loss during the period | |
| (47,413 | ) | |
| 110,313 | |
| Other prior year (gain)/loss recognized in period | |
| (157,632 | ) | |
| - | |
| Effect of settlement, curtailment | |
| (58,787 | ) | |
| - | |
| Amortization of pension related net loss | |
| - | | |
| - | |
| Net (gain)/loss, end of period | |
| (105,759 | ) | |
| 161,104 | |
| | |
| | | |
| | |
| Prior service cost, beginning of period | |
| - | | |
| - | |
| Amortization of prior service cost | |
| (3,094 | ) | |
| (3,031 | ) |
| Prior service cost end of period | |
| (3,094 | ) | |
| (3,031 | ) |
| | |
| | | |
| | |
| Total unrecognized pension (credit) cost, end of period | |
$ | (108,853 | ) | |
$ | 158,073 | |
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The weighted average of the key assumptions used
to compute the benefit obligations were as follows:
| | |
2024 | | |
2023 | |
| Discount rate | |
| 0.95 | % | |
| 1.45 | % |
| Rate of increase in compensation level | |
| 0.65 | % | |
| 0.65 | % |
| Interest credit rate on savings accounts | |
| 1.25 | % | |
| 1.45 | % |
| Expected long-term rate of return on plan assets | |
| 2.15 | % | |
| 2.30 | % |
| Inflation rate | |
| 1.25 | % | |
| 1.25 | % |
The assumption of the expected long-term rate
of return on plan assets was based on the long-term historical rates of returns for the different investment categories which were adjusted,
where appropriate, to reflect financial market developments. The investment risk is borne by the insurer and the reinsurer respectively,
and the investment decision is taken by the board of trustees of the collective insurance. The accumulated benefit obligation as of December
31, 2024, and 2023 amounted to $2,213,177 and $809,028, respectively.
Note 9
Commitments and Contingencies:
Commitments
On March 10, 2021, the Company entered into a
license agreement with Novartis Pharma AG (“Novartis”), whereby the Company obtained, on an exclusive basis in the U.S., all
of the available data referred to and included in the original new drug application (“NDA”) for Sanorex® (mazindol) submitted
to the U.S. Food and Drug Administration (“FDA”) in February 1972. The agreement encompasses all preclinical and clinical
studies, data used for manufacturing including stability and other chemistry manufacturing and controls data, formulation data and know-how
for all products containing mazindol as an active substance, and all post-marketing clinical studies and periodic safety reports from
1973 onwards. Under the agreement, the Company has obtained the same rights on a non-exclusive basis in all territories outside of the
U.S. except for Japan, with the right to cross-reference the Sanorex NDA with non-U.S. regulatory agencies in the licensed territories.
The Agreement includes the right to sublicense or assign the license to third parties, subject to such third parties meeting certain obligations.
As consideration for the license, the Company paid Novartis $250,000 upon the signing of the agreement with milestone payments due as
follows: (i) $750,000 payable following the end of a Phase II meeting with the FDA, with the amount to be reduced to $375,000 if toxicology
studies must be repeated; (ii) $2 million following the earlier of U.S. Food and Drug Administration (“FDA”) marketing authorization
of Quilience or Nolazol; (iii) 1% of any upfront and milestone payments, if any, from any sublicensees and (iv) $3 million as a one-time
payment upon the Company’s product candidate reaching $250 million in cumulative sales.
Litigation
The Company may become involved in miscellaneous
litigation and legal actions, including product liability, consumer, commercial, tax and governmental matters, which can arise from time
to time in the ordinary course of the Company’s business. Litigation and legal actions are inherently unpredictable, and excessive
verdicts can result in such situations.
On December 1, 2023, the Company received a
letter from Cambrex Corporation, stating that as of December 1, 2023, the Company has an overdue balance for services completed
under certain proposals by and between the Company, Cambrex High Point, Inc. and Avista Pharma Solutions, Inc. in the aggregate
amount of $492,723. On October 10, 2024, the Company successfully settled the outstanding claim through a debt purchase agreement
with an accredited investor, in the amount of $200,013, which effectively resolved the creditor claim.
On August 21, 2024, the Company received a letter
from Dunn Lambert LLC, the law firm representing Clinilabs, Inc., or Clinilabs, stating that a complaint had been filed in the Superior
Court of New Jersey. The complaint concerns three unpaid invoices totaling $886,412 plus interest at a rate of 6%. On June 1, 2023,
Clinilabs entered into a start-up agreement with the Company. On December 4, 2023, the Company received five credit notes and two invoices
from Clinilabs for services performed under the start-up agreement. On October 10, 2024, the Company successfully settled the outstanding
claim through a debt purchase agreement with an accredited investor, in consideration of $500,223 and 49,475 common shares equal in
value to $279,300, which effectively resolved the creditor claim.
On December 11, 2023, the Company received a notice
alleging several causes of action, including a failure to remit payment for services rendered by CoreRX. On October 10, 2024, the Company
successfully settled the outstanding claim through a debt purchase agreement with an accredited investor, in the amount of $511,835, which
effectively resolved the creditor claim.
On June 11, 2024, the Company received a civil
court complaint filed in the Civil Court of the City of New York by Chad Hellman, the former Chief Financial Officer of the Company. The
Claimant is demanding approximately $35,171 for unpaid consulting fees. The Company responded to the civil court complaint in July 2024.
On October 3, 2024, Chad Hellman dismissed his claim. The Company successfully settled the outstanding claim through payment of $36,940
to Chad Hellman on October 7, 2024.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
On August 27, 2024, the Company received correspondence
from Université de Lausanne, initiating the official “audience de conciliation” procedure, overseen by the ordinary
civil court in Lausanne. The hearing was scheduled for October 9, 2024, at the Tribunal d’arrondissement de Lausanne. The complaint
pertains to an unpaid invoice for research services amounting to $110,179, plus interest at a rate of 5%. At the hearing on October 9,
2024, Université de Lausanne was not open to discussing a potential settlement. The Company asserts that the services provided
did not meet the required standard of care and intends to defend its position. On 9 May 2025, the Company filed a statement of defense
and asserted a counterclaim in CHF 30,000 plus compensatory interests at a rate of 5% per annum since June 29, 2022.
Note 10
Equity:
Preferred Shares
In October 2024, the Company amended and restated
its Articles of Association to designate 806,452 of preferred shares with a par value of 0.80 CHF in connection with the execution of
a securities purchase agreement. The preferred shares have the following rights, preferences and privileges:
| ● | Accrue dividends at an annual rate of eight percent
(8%) of the stated value of the preferred shares from the date of issuance; |
| | | |
| ● | Stated value and initial conversion price of
$4.96, subject to adjustment, for stock splits, dividends and subsequent equity sales and issuances below the initial conversion price;
|
| | | |
Upon any liquidation, dissolution or winding up of the Company, the
preferred shareholders will be entitled to receive an amount equal to the stated value of any preferred shares held at the time of such
an event prior to any holders of common shares.
On October 9, 2024, the Company entered into a securities purchase
agreement, or the Debt Purchase Agreement, with an accredited investor, pursuant to which in exchange for the satisfaction of the Company’s
debt in the aggregate amount of $4.0 million held by the investor, the Company agreed to issue 806,452 newly designated convertible preferred
shares, at a purchase price of $4.96 (rounded). The preferred shares contain an initial conversion price of $4.96 per share. The transactions
contemplated by the Debt Purchase Agreement closed on October 10, 2024. Pursuant to the Debt Purchase Agreement, the Company agreed to
grant the investor the right to purchase up to an additional $10.0 million worth of convertible preferred shares beginning six months
after the closing and continuing for as long as the investor owns preferred shares. Any additional preferred shares issued upon the investor’s
right being exercised will be identical to the initial preferred shares except the conversion price will be based on the average daily
closing sale price of common shares for the five trading days prior to the investor giving notice of its intent to exercise its rights.
Additionally, pursuant to the Debt Purchase Agreement, the Company agreed to grant the investor the right to participate in up to fifty
percent (50%) of future offerings of the Company’s securities for one year following the closing. In addition, the Company agreed
not to enter into an equity line of credit or similar agreement, without the consent of the majority of the holders of the preferred shares.
In December 2024, 600,000 preferred shares were converted into Company Common Shares; therefore, as of December 31, 2024 206,452 preferred
shares remain outstanding. Accrued dividends from the date of issuance to December 31, 2024 were not significant.
Common Shares
As of December 31, 2024, the Company had 3,159,535
registered and issued Company Common Shares.
On December 4, 2024, the Company, entered into a securities purchase agreement, or the December
SPA, with a certain accredited investor and agreed to issue and sell to the investor, in a private placement offering, or the SPA Offering,
up to 322,580 Company Common Shares, at a purchase price of $3.10 per Company Common Share for aggregate gross proceeds of up to $1 million,
subject to shareholder approval, or the Shareholder Approval.
The offering was structured in two tranches, with
the first tranche of $500,000 closing following shareholder approval. The Company has also agreed that (i) on or before January 15, 2025,
or sooner contemporaneously with the Shareholder Approval, the Company will take all steps necessary to obtain all approvals, including
shareholder approval, necessary to reduce the par value of the Company Common Shares to the minimum permissible par value which shall
not be greater than CHF 0.03 per Company Common Share; (ii) on or before January 9, 2025, the Company will authorize and reserve sufficient
Company Common Shares to satisfy the anti-dilution and ratchet rights of the Purchaser under the prior securities purchase agreement dated
October 9, 2024, or the October SPA, after giving effect to the conversion price reduction of the preferred shares as a result of the
Dilutive Issuances (as defined in the October SPA) caused by the Company entering into Securities Purchase Agreement; and (iii) in addition
to the Company’s obligations pursuant to the October SPA. The second tranche of $500,000 may occur, at the election of the investor,
within 15 days following the Company meeting certain conditions, including the receipt of shareholder approval and the Common Shares trading
for at least ten consecutive trading days above the purchase price of $3.10, which corresponds to an approximate 15% premium. As of December
31, 2024, the conditions for closing have not yet occurred. See Note 13 for further details.
In addition, the Company has agreed to register
the Company Common Shares subject to the terms of the previously executed registration rights agreement dated October 9, 2024, or the
Registration Rights Agreement, such that the Company Common Shares must be registered within 30 days of the closings.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
On October 10, 2024, the Company
successfully implemented a crucial restructuring measure by converting the claims of debt holders in the amount of $2,788,650 into
493,986 Company Common Shares, (“Debt Conversion”) This conversion was facilitated through an ordinary capital increase,
providing the necessary shares for the debt holders. This conversion was facilitated through an ordinary capital increase, providing
the necessary shares for the debt holders, and was recorded at fair market value with no gain or loss recognized in the statement of
operations.
On October 9, 2024, the Company entered into the
October SPA, with certain accredited investors and agreed to issue and sell to the investors, in a private placement offering, (i) 806,452
Company Common Shares, and (ii) common share purchase warrants, or the Common Warrants, to purchase 806,452 Company Common Shares, at
a combined purchase price of $3.97, for aggregate gross proceeds of $3.2 million. The Common Warrants have a term of five years and have
an exercise price of $4.25 per share. Pursuant to the October SPA, the Company agreed to grant the investors the right to participate,
in the aggregate, in up to fifty percent (50%) of future offerings for one year following the closing of the offering. In addition, the
Company agreed not to enter into an equity line of credit or similar agreement, without the consent of the majority of the holders of
the preferred shares. The transactions contemplated by the October SPA closed on October 10, 2024.
On June 28, 2024, the Company entered into a securities
purchase agreement for the issuance of 81,944 Company Common Shares at a purchase price of $9.60 per share. The offering
closed on July 1, 2024. Investors also received unregistered warrants to purchase up to 81,944 Company Common Shares at an exercise
price of $9.60 per share. These warrants are exercisable upon issuance and will expire five years from the date of issuance.
The offering resulted in gross proceeds of $786,660. The net proceeds were used for working capital and general corporate purposes.
H.C. Wainwright & Co., LLC served as the
exclusive placement agent for the offering, or the Placement Agent. The Company agreed to pay the Placement Agent a cash fee of 7.0%
of the gross proceeds, a management fee of 1.0% of the gross proceeds, and $50,000 for accountable expenses. Additionally,
the Company issued to the Placement Agent warrants to purchase up to 5,736 Company Common Shares at an exercise price of $12.00 per
share. These warrants are exercisable from the date of issuance until five years from the commencement of sales in the offering.
On March 20, 2024, the Company entered into a
securities purchase agreement, or the Purchase Agreement, providing for the issuance in a registered direct offering of 175,000 Company
Common Shares at a purchase price of $10.00 per share that closed on March 22, 2024. In addition, pursuant to the Purchase Agreement,
the investors received unregistered warrants, or the Common Warrants, to purchase up to an aggregate of 87,500 Company Common Shares
at an exercise of $10.00 per share in a concurrent private placement. The Common Warrants were immediately exercisable upon issuance
and will expire five years following the date of issuance. The Company has also agreed that from the date of the Purchase Agreement until
one year after the closing date of the offering, the Company shall not enter into an agreement to effect any issuance by the Company
or any of the Company’s subsidiaries of Company Common Shares or common share equivalents (or a combination of units thereof) involving
a variable rate transaction. The offering resulted in gross proceeds to the Company of $1,750,000 and expenses of approximately $321,000.
Warrants
On October 9, 2024, the Company and certain existing
warrant holders entered into warrant amendment agreements, or collectively, the Amendment, to amend those warrants issued by Company to
such holders, collectively, to purchase up to 105,843 Common Shares issued to such holders, or the Existing Warrants. The Amendment makes
certain adjustment to the definition of a “Fundamental Transaction” in Section 3(e) of the Existing Warrants. In exchange
for the Amendment, the Company agreed to adjust the exercise price in the Common Warrants to CHF 0.80 and issued to the holders Pre-Funded
Warrants to purchase up to 136,648 Common Shares, or the Pre-Funded Warrants. Each Pre-Funded Warrant is exercisable for one Common Share
at an exercise price of CHF 0.80 per share. The Pre-Funded Warrants are immediately exercisable and may be exercised at any time until
all of the Pre-Funded Warrants are exercised in full.
On September 16, 2024, the Company and an institutional
investor entered into a warrant amendment agreement to amend warrants to purchase up to 172,836 common shares, par value CHF 0.80. The
amendment adjusted the definition of a “Fundamental Transaction” and the exercise price to CHF 0.80. Following an increase
in authorized common shares, the Company will issue pre-funded warrants to purchase up to 191,431 common shares for aggregate proceeds
of $153,144.
The Company has reflected a deemed dividend totaling $2,076,180
for the incremental value from the modification of the warrants’ exercise price and the fair value of the warrants issued as consideration
for executing the Amendment. The deemed has been reflected within stockholders’ deficit and as an increase to loss attributable
to common stockholders for the purposes of computing loss per share.
The incremental value due to the modification
was determined to be $366,684 using a Black-Scholes option pricing model to compute the fair value of the warrant pre and post modification
and recording the difference.
The fair value of the aggregate warrants issued
as consideration for executing the Amendment was determined to be $1,709,496 computed using a Black-Scholes option pricing model.
The following input assumptions were utilized
in the Black-Scholes model: expected term of one year, volatility of 148%, risk-free rates ranging from 3.96% to 4.24%, and dividend rate
of 0%.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The following table summarizes the common share
warrant activity for the year ended December 31, 2024:
| Balance at January 1, 2024 | |
| 476,128 | |
| Issuances | |
| 1,321,961 | |
| Exercises | |
| (191,431 | ) |
| Expired | |
| (7,696 | ) |
| Balance at December 31, 2024 | |
| 1,598,962 | |
The intrinsic value of exercisable but unexercised
in-the-money common share warrants at December 31, 2024 was $344,802.
Option Plan
On December 14, 2021, the Board of Directors adopted
the Share Option Plan Regulation 2021 (the “Option Plan”). The purpose of the Option Plan is to retain, attract and motivate
management, employees, directors and consultants by providing them with options to purchase the Company’s common shares. The Board
of Directors allocated fifteen percent (15%) of the Company’s fully diluted shares to awards that may be made pursuant to the Option
Plan.
The exercise prices, vesting and other restrictions
of the awards to be granted under the Option Plan are determined by the Board of Directors, except that no stock option may be issued
with an exercise price less than the fair market value of the common shares at the date of the grant or have a term in excess of ten years.
Options granted under the Option Plan are exercisable in whole or in part at any time subsequent to vesting.
The following table summarizes total stock option
activity for the year ended December 31, 2024:
| | |
Number of
Options | | |
Weighted
Average
Exercise
Price | |
| Balance at December 31, 2022 | |
| 33,328 | | |
$ | 49.20 | |
| Granted | |
| 19,409 | | |
| 30.40 | |
| Exercised | |
| - | | |
| | |
| Expired/cancelled | |
| (6,145 | ) | |
| 39.60 | |
| Balance at December 31, 2023 | |
| 46,591 | | |
$ | 42.74 | |
| Granted | |
| - | | |
| - | |
| Exercised | |
| - | | |
| - | |
| Expired/cancelled | |
| (12,174 | ) | |
$ | 30.31 | |
| Balance at December 31, 2024 | |
| 34,417 | | |
$ | 47.14 | |
| Options vested and exercisable at December 31, 2024 | |
| 19,116 | | |
$ | 51.95 | |
The weighted average remaining contractual life of each of the options
outstanding at December 31, 2024 was 7.62 years.
Stock-based compensation expense was $125,644, $164,906, and $22,730
for the years ended December 31, 2024, 2023, and 2022, respectively, and was recorded in general and administrative expenses. As of December
31, 2024, total unrecognized stock-based compensation expense relating to unvested stock options was $117,119. This amount is expected
to be recognized over a weighted-average period of 2 years.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The following table summarizes unvested stock option activity for the
year ended December 31, 2024:
| | |
Non-Vested
Options | | |
Weighted
Average
Grant date
Fair Value | |
| | |
| | |
| |
| Balance at December 31, 2022 | |
| 32,078 | | |
$ | 10.00 | |
| Granted | |
| 19,409 | | |
| 20.40 | |
| Vested | |
| (9,988 | ) | |
| 9.60 | |
| Expired/cancelled | |
| (6,145 | ) | |
| 10.80 | |
| Balance at December 31, 2023 | |
| 35,354 | | |
$ | 15.60 | |
| Granted | |
| - | | |
| | |
| Vested | |
| (7,879 | ) | |
| 53.41 | |
| Expired/cancelled | |
| (12,174 | ) | |
| 30.31 | |
| Balance at December 31, 2024 | |
| 15,301 | | |
$ | 41.11 | |
The aggregate intrinsic value of stock options
is calculated as the difference between the exercise price of the stock options and the fair value of the Company’s common shares
for those stock options that had exercise prices lower than the fair value of the Company’s common shares. The share price as of
December 31, 2024, was $2.11 and the aggregate intrinsic value for options outstanding and expected to vest each year was nil. The intrinsic
value of exercisable options was nil as the exercise price was greater than the share price.
Note 11
Income Taxes:
The Company has Swiss tax loss carryforwards of $52.2 million as of
December 31, 2024 (December 31, 2023: $47.2 million) of which $32.9 million will expire within the next five years, and $14.3 million
will expire between 2030 - 2031.
The significant components of net deferred taxes
as of December 31, 2024, and 2023 are shown in the following table:
| | |
2024 | | |
2023 | |
| Deferred tax assets: | |
| | |
| |
| Net benefit from tax loss carryforwards | |
$ | 5,536,408 | | |
$ | 5,000,743 | |
| Deferred revenues | |
| - | | |
| 264,997 | |
| | |
| | | |
| | |
| Valuation allowance | |
| (5,536,408 | ) | |
| (5,265,740 | ) |
| | |
| | | |
| | |
| Net deferred taxes | |
$ | - | | |
$ | - | |
The Company recorded a valuation allowance in
2024 and 2023 to reduce the net deferred taxes, as the Company deemed it to be more likely than not that the future deferred tax assets
would not be realized in the future based on the lack of sufficient positive evidence in the jurisdictions related to the realization
of the deferred tax assets.
The effective tax rate was 0% for the years ended
December 31, 2024, 2023 and 2022. The following table shows the income taxes in 2024, 2023 and 2022:
| | |
2024 | | |
2023 | | |
2022 | |
| | |
| | |
| | |
| |
| Current tax | |
$ | - | | |
$ | - | | |
$ | - | |
| Deferred income tax (benefit) | |
| (270,668 | ) | |
| (1,515,501 | ) | |
| (1,004,681 | ) |
| | |
| (270,668 | ) | |
| (1,515,501 | ) | |
| (1,004,681 | ) |
| Change in valuation allowance | |
| 270,668 | | |
| 1,515,501 | | |
| 1,004,681 | |
| Total income tax expense | |
$ | - | | |
$ | - | | |
$ | - | |
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The Company files income tax returns in Switzerland.
The Company’s income tax position in Switzerland is finally assessed up to the year ended December 31, 2020, so the years ended
December 31, 2021, 2022, 2023 and 2024 are open for examination. Currently the Company does not have any open tax assessments. The following
table shows the reconciliation between expected and effective tax rate:
| | |
2024 | | |
2023 | | |
2022 | |
| | |
| | |
| | |
| |
| Statutory tax rate | |
| 10.6 | % | |
| 10.6 | % | |
| 10.6 | % |
| Effect of temporary differences | |
| 3.1 | % | |
| 1.9 | % | |
| (4.8 | )% |
| Change in valuation allowance on deferred tax assets | |
| (13.7 | )% | |
| (12.5 | )% | |
| (5.8 | )% |
| | |
| | | |
| | | |
| | |
| Effective tax rate | |
| - | % | |
| - | % | |
| - | % |
The Company had generated approximately $9.7 million
of net operating losses (“NOLs”) prior to the reorganization in 2019 in which the Company’s preliminary analysis indicates
that such NOLs would not be subject to significant limitations pursuant to applicable income tax regulations. The temporary differences
relate to book to tax differences from the adjustments required to present the consolidated financial statements on a U.S. GAAP basis
from local Swiss GAAP, mainly in the areas of debt, intangible amortization and deferring financing costs.
As of December 31, 2024, 2023, and 2022,
there were no unrecognized tax benefits. If such matters were to arise, the Company would recognize interest and penalties related
to income tax matters in income tax expense. The Company did not incur any material interest or penalties in connection with income
taxes during the years ended December 31, 2024, 2023 and 2022.
Note 12
Related party consulting agreements:
The Company entered into consulting agreements
with some of its senior management.
In January 2017, and as subsequently amended in October 2020, the Company entered into a consulting agreement with CHG BioVenture SA,
an entity controlled by Mr. Hervé Girsault, the Company’s former Head of Business Development. On May 1, 2021, for the continuation
of Mr. Girsault’s engagement with the Company in his then current role. The Company agreed to pay CHG BioVenture SA a monthly fee
of CHF 4,375 ($4,733) plus VAT for its services. In addition, CHG BioVenture SA was eligible for a 1% success fee payment in the
event of closing of a partnering agreement in China. The consulting agreement was terminated in October 2024 and provides for a 24-month
non-competition clause along with standard confidentiality provisions. For the years ended December 31, 2024, 2023 and 2022, the Company
recorded fees to CHG BioVenture SA of $79,454, $121,658 and $131,941, respectively, included in general and administrative expenses on
the statement of operations and comprehensive loss.
In February 2021, and as amended in July 2021,
the Company entered into a consulting agreement with Mr. Eric Konofal, the Company’s current Chief Scientific Officer, pursuant
to which the Company agreed to pay Mr. Konofal a daily rate of CHF 2,000 for his services. The consulting agreement may be terminated
by either party upon 30 days’ written notice or immediately by the Company in the event of a material breach by Mr. Konofal that
cannot be cured. The consulting agreement contains customary confidentiality provisions and provides for an 18-month non-solicitation
clause as well as reimbursement for certain expenses. For the years ended December 31, 2024, 2023 and 2022, the Company recorded fees
to Mr. Konofal of $107,700, $178,820 and $201,053, respectively, included in research and development expenses on the statement of operating
and comprehensive loss.
On March 19, 2024, the Company entered into an
exclusive license agreement (the “Aexon Agreement”), with Aexon Labs Inc., a Delaware corporation, or Aexon. Alexander Zwyer,
(the Company’s Chief Executive Officer) owns 35% of Aexon, and Eric Konofal, (the Company’s Chief Scientific Officer) owns
59% of Aexon. Mr. Konofal is the founder of Aexon, with which the Company has a license agreement, and also serves as the President of
Aexon. Mr. Zwyer holds no board or executive position at Aexon Labs. Pursuant to the Aexon Agreement, Aexon granted the Company an exclusive,
royalty-bearing license (“License”), with the right to grant sublicenses in multiple tiers according to the terms of the Aexon
Agreement. Subject to earlier termination of the Aexon Agreement in accordance with its terms, the term of the Aexon Agreement is from
the effective date of the Aexon Agreement to the latest of (i) the Company’s termination of the commercialization of one or more
pharmaceutical or therapeutic products, or any combination thereof, in the use of such compounds for narcolepsy and other neuro degenerative
disorders in the last region and country in which commercialization had actually begun, and (ii) the expiration of the last-to-expire
Valid Claim (as defined in the Aexon Agreement) of a patent identified in the Aexon Agreement and patents owned by Aexon as of the date
of the Aexon Agreement, that covers such pharmaceutical or therapeutic product for the use of such compounds for narcolepsy and other
neuro degenerative disorders in the respective country or region in which it was used. Pursuant to the terms of the Aexon Agreement, the
Company agreed to pay Aexon a royalty on a country-by-country basis of 5% to 30% depending on (i) earnings by the Company in a specified
region or country for licensed products covered by patents, (ii) whether the applicable patent has not been granted to the applicable
product at the time of commercialization of such product and (iii) whether the Company challenges the validity of a patent.
NLS PHARMACEUTICS LTD. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
The Company made an upfront payment of $30,000
for the option exclusivity and $170,000 upon execution of the definitive agreement to exercise the option for the Aexon Agreement, of
which $40,000 was paid in cash and the remaining $130,000 was paid in 23,028 common shares. In addition, Aexon will receive 15% of all
proceeds earned by the Company in any future sub-licensing agreements. The Company must also make royalty payments to Aexon upon
the occurrence of certain milestones. Such payments upon the occurrence of milestones contemplated in the Aexon Agreement range from $100,000
to $300,000, in addition to percentage royalty payments ranging from 5% to 15% which may decrease or increase up to 30% if the Company
challenges the validity of the patents under the agreement, depending on the result of such challenge. Further, pursuant to the Aexon
Agreement, the Company has agreed to pay Aexon a percentage of license fees, milestones and royalties received from sublicensees, ranging
between 5% and 15% which may decrease or increase up to 30% if the Company challenges the validity of the patents under the agreement,
depending on the result of such challenge. For the year ended December 31, 2024, the Company recorded fees to Aexon of $170,000 which
is included in research and development expenses on the statement of operating and comprehensive loss.
In October 2024, the Company entered into a consulting
agreement with Ms. Nicole Fernandez-McGovern, the Company’s current Chief Financial Officer, pursuant to which the Company agreed
to pay Mr. Fernandez-McGovern a monthly retainer for her services of $18,000 per month. For the year ended December 31, 2024, the Company
recorded fees to Ms. Fernandez-McGovern of $54,316 which is included in general and administrative expenses on the statement of operating
and comprehensive loss.
Note 13
Subsequent Events:
Management has evaluated subsequent events that
have occurred through the date these consolidated financial statements were issued.
In January 2025, the Company completed the initial
closing of $500,000 of the previously announced $1 million financing between the Company and an accredited investor under a securities
purchase agreement dated December 4, 2024, with the approval of certain items in Company’s extraordinary general meeting on January
7, 2025. As of May 15, 2025, the conditions for the second tranche were not met and did not occur.
On January 30, 2025, the Merger Agreement was
amended such that it includes the right of the Company and Kadimastem to terminate the Merger Agreement if the Closing shall not have
occurred on or before April 30, 2025, which outside date can be further extended by mutual agreement. Further, the amendment included
an additional provision providing that any shareholder receiving common shares in excess of a 9.99% beneficial ownership limitation as
a result of the Merger, shall instead be issued pre-funded warrants exercisable for a number of common shares in excess of the beneficial
ownership limitation, at an exercise price equal to the par value of the common shares as of the Effective Time, which, in any event,
shall be no less than CHF 0.0001 per share. In addition, on May 5, 2025, the Merger Agreement was further amended to further extend the
outside date to June 30, 2025.
On March 27, 2025, the Company entered into a
securities purchase agreement, or the March 2025 SPA, with three accredited investors. Pursuant to the terms of the March 2025 SPA, the
Company agreed to issue and sell to the investors, in a private placement offering, or the March 2025 Offering, 1,212,122 Preferred Shares
with a conversion price of $1.65 per share, for aggregate gross proceeds of $2 million. Pursuant to the terms of the March 2025 SPA, the
investors may purchase up to $1 million of additional Preferred Shares on identical terms as the initial closing, subject to the Company
obtaining shareholder approval. The March 2025 Offering closed on March 28, 2025.
The initial closing of the March 2025 Offering
resulted in gross proceeds to the Company of to $2 million. The Company intends to use the net proceeds from the March 2025 Offering for
working capital and general corporate purposes, including for expenses relating to the Merger.
Further, on March 31, 2025, the Company entered
into a Common Shares Purchase Agreement with Alpha Capital Anstalt, or the Facility SPA, relating to a committed equity facility. Pursuant
to the Facility SPA, the Company has the right from time to time at its option to sell to Alpha up to $25.0 million of Company Common
Shares, subject to certain conditions and limitations set forth in the Facility SPA.
The purchase price of the Company Common
Shares that the Company elects to sell to Alpha pursuant to the Facility SPA will be 95% of the volume weighted average price of the
Company Common Shares during the applicable purchase date on which the Company has timely delivered written notice to Alpha
directing it to purchase Company Common Shares under the Facility SPA.
In connection with the execution of the Facility
SPA, the Company agreed to issue a pre-funded warrant, or the Pre-Funded Warrant, to purchase $250,000 in Company Common Shares to Alpha
as consideration for its irrevocable commitment to purchase the Company Common Shares upon the terms and subject to the satisfaction of
the conditions set forth in the Facility SPA.
F-28